

In my experience working with quality teams, GxP compliance in pharma is often perceived as a burden. However, it's a framework that protects what matters most to our industry: patients, product quality, and regulators' trust.
Every stage of the journey, from the first experiments in the lab to commercial manufacturing, global distribution, and post-market monitoring, is shaped by GxP standards.
What’s changed is the level of complexity.
Supply chains now span continents, regulations evolve faster than ever, and digital systems are replacing the binders and paper trails we used to rely on. The days of managing compliance with stacks of SOPs on a shelf are gone.
Today, validated systems, secure audit trails, and digital quality platforms are essential to prove that processes are controlled and records are trustworthy.
This guide is designed to give you clarity. I'll explore key concepts such as:- What is GxP in pharma?
- GxP vs GMP
- What is GxP and non gxp in pharma?
I’ll also share how technology, like eQMS platforms and digital validation, can transform compliance from a reactive into a proactive advantage, helping teams stay inspection-ready while gaining efficiency and resilience.
By the end, you’ll understand why GxP compliance in pharma is more than a box to tick - let’s go!
What is GxP in pharma?
Before we dive into specific regulations for GxP compliance in pharma, let’s start with the basics: what GxP actually means in a pharmaceutical context. So, what is GxP in pharma?
In simple terms, GxP in pharma is a set of international quality guidelines that ensure medicines and medical devices are developed, produced, and managed in a way that guarantees patient safety, product quality, and data integrity.
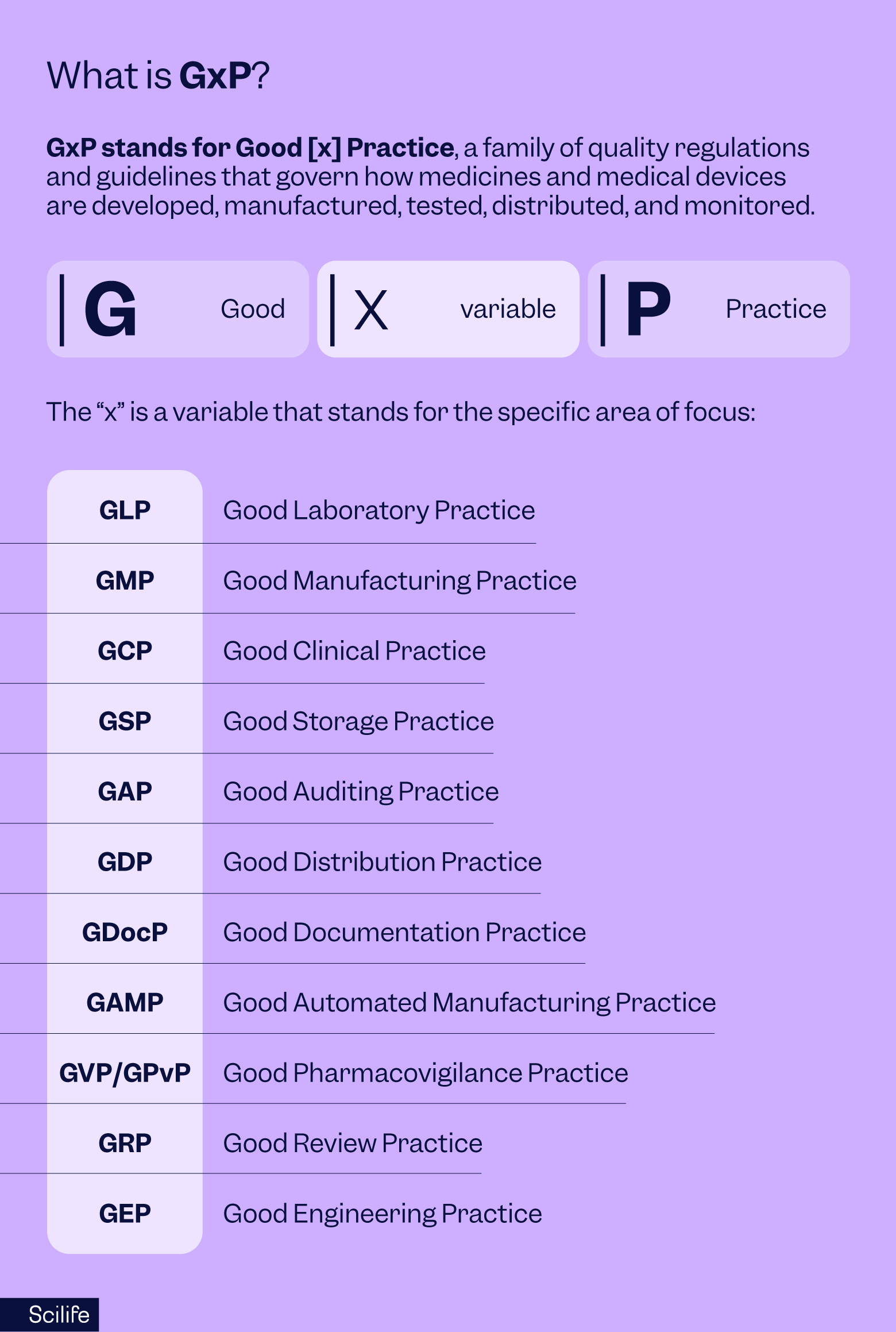
GxP stands for Good [x] Practice, a family of quality regulations and guidelines that govern how medicines and medical devices are developed, manufactured, tested, distributed, and monitored.
Unlike a single regulation or certification, GxP is a broad umbrella of practices. Together, these practices create a regulatory framework that ensures medicines are safe, effective, and consistently high in quality.
- G = Good
- x = Variable (L, M, C, D, etc.)
- P = Practice
The “x” is a variable that stands for the specific area of focus:
- GLP - Good Laboratory Practice
- GMP -Good Manufacturing Practice
- GCP - Good Clinical Practice
- GSP - Good Storage Practice
- GAP - Good Auditing Practice
- GDP- Good Distribution Practice
- GDocP - Good Documentation Practice
- GAMP - Good Automated Manufacturing Practice
- GVP/GPvP - Good Pharmacovigilance Practice
- GRP - Good Review Practice
- GEP - Good Engineering Practice
Each company must determine which GxPs apply to its activities. For example, a CRO running toxicology studies must comply with GLP, while a pharmaceutical manufacturer must comply with GMP. A biotech in clinical development will need both GCP and GMP.
The origins of GxP trace back to GMP regulations first introduced by the FDA in 1963, largely in response to drug safety tragedies like the thalidomide crisis. Over time, additional “Good Practices” were added, creating the GxP family we know today.
In practice, GxP represents the minimum global standard for market access. It guarantees that medicines are developed, tested, produced, stored, and distributed under controlled, traceable, and auditable conditions. Without it, no product can move from the laboratory bench to the patient’s bedside.
Now that we know what GxP covers, the next question is: who carries the responsibility of GxP?
The answer is simple: everyone.
Scilife Tip:
Anyone wondering what is GxP in pharma should see it not as paperwork, but as the safety net that keeps medicines effective and trustworthy across every stage of development and distribution.

Who is responsible for ensuring GxP quality?
Quality is everyone’s responsibility. GxP regulations in pharma cannot be delegated to a single department; it must be embedded throughout the organization.
Regulators in both the US and Europe emphasize that GxP compliance is a shared responsibility across every role. From senior executives to operators, from scientists to IT administrators, every role in the product lifecycle contributes to adherence to GxP regulations in pharma.
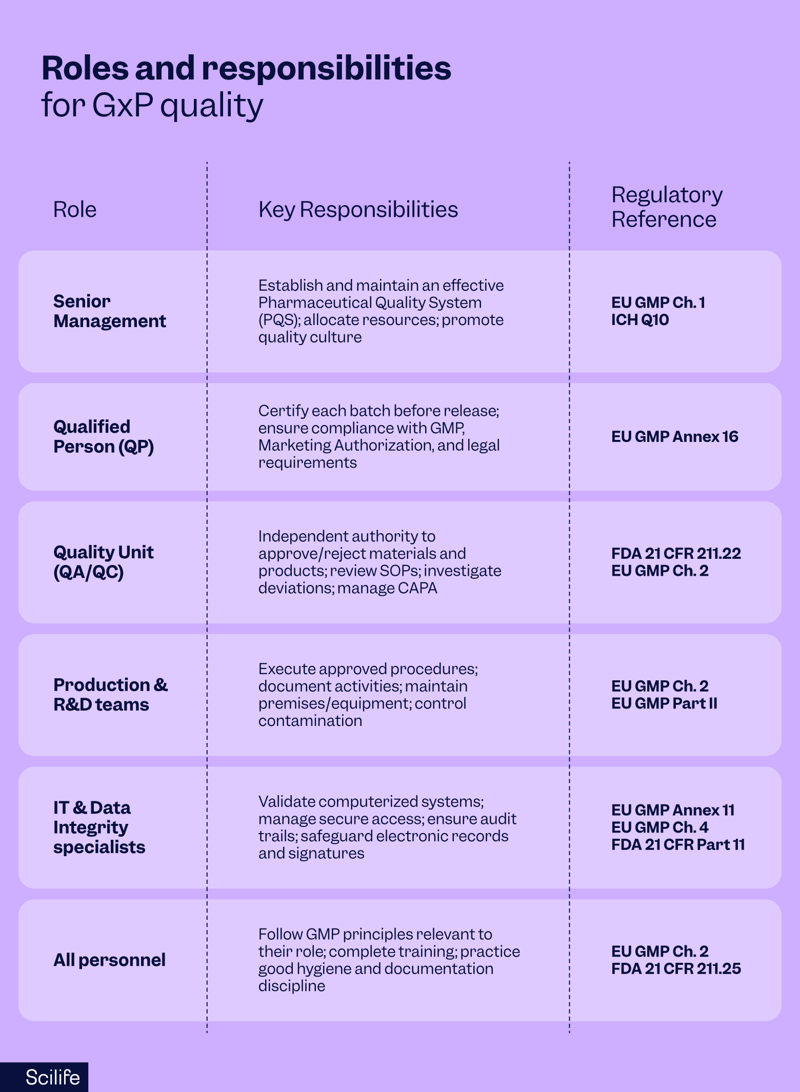
At the top, senior management bears ultimate accountability. EU GMP Chapter 1 states that leadership is responsible for establishing and maintaining an effective Pharmaceutical Quality System (PQS). Management must ensure sufficient resources, training, and a culture that prioritizes quality over speed or cost.
In Europe, Qualified Persons (QPs) carry a unique legal obligation. According to Annex 16 of the EU GMP, no batch of medicinal product can be released to the market without certification by a QP, who must personally ensure compliance with GMP, the Marketing Authorization, and all applicable laws. This accountability is individual and cannot be transferred.
In the US, Quality Units (QA/QC) hold similar authority. Under 21 CFR 211.22, the Quality Control Unit is empowered to approve or reject all materials, components, and finished products. QA/QC also oversees SOPs, deviation investigations, and Corrective and Preventive Actions (CAPAs), ensuring independence from production.
Meanwhile, Production and R&D teams must follow approved procedures, document their activities, and maintain premises and equipment to prevent contamination or errors.
For active substances, EU GMP Part II extends these duties to raw material handling, calibration, and contamination control.
With the rise of digital systems, IT and data integrity specialists have become equally important. EU GMP Annex 11 and FDA 21 CFR Part 11 mandate validated systems, secure access, audit trails, and electronic records management. If systems fail, compliance fails, making IT a frontline quality function.
Finally, all employees have a responsibility to understand how their role impacts product quality. EU GMP Chapter 2 emphasizes that staff must be trained, qualified, and continuously updated on the procedures relevant to their work.
Compliance is not the responsibility of the quality team alone; it is a shared effort where every role contributes to patient safety and trust. Scilife supports this collective approach by making quality processes visible, accessible, and easy to follow across the organization, helping teams sustain GxP regulations over time.
Without this culture, GxP compliance in pharma cannot be sustained.
Because GMP is the most familiar of the GxPs, it’s often confused with the whole concept of GxP. Let’s clarify the distinction.
GxP vs GMP: What’s the difference?
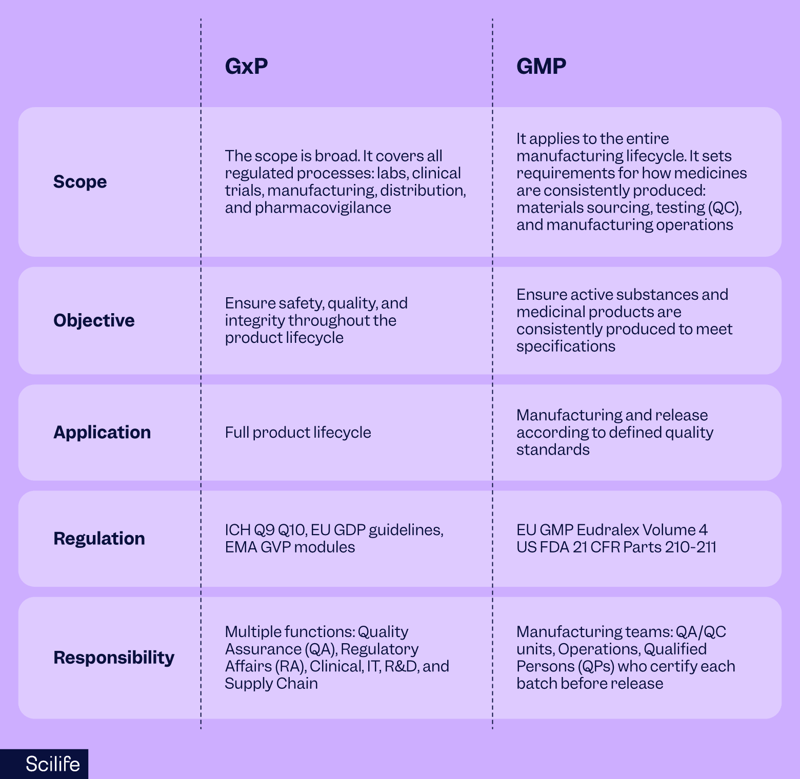
When comparing GxP vs GMP, it’s important to understand the scope difference.
GxP is the umbrella framework that covers all Good [x] Practices, while GMP is one specific part of that framework.
GxP is the umbrella framework covering all the “Good Practices” across the product lifecycle: GLP for labs, GCP for trials, GDP for logistics, GVP for safety monitoring, and more.
GMP is one specific subset, focused solely on manufacturing. It ensures medicines are consistently produced and controlled to the required quality standards, covering raw materials, facilities, equipment, training, documentation, and batch release.
In the US, GMP requirements are found in 21 CFR Parts 210–211; in the EU, in EudraLex Volume 4.
All GMP is GxP, but not all GxP is GMP. GMP focuses only on manufacturing processes, but GxP compliance in pharma extends far beyond production. That's GxP vs GMP for you in a nutshell!
Understanding the difference between GxP and GMP sets the stage for an even bigger question: why is GxP important in pharma?
Why are GxP regulations important in pharma?
GxP regulations in pharma are the foundation of patient safety, product quality, and regulatory trust. Without it, no medicinal product can legally reach patients.
Compliance is not just about meeting a checklist of requirements; it is what ensures every pill, vial, or injection patients receive is safe, effective, and manufactured under controlled conditions.
Protecting product quality and patient safety
The primary goal of GxP is to protect patients from harm. By embedding controlled processes and validated systems into every stage of the product lifecycle, companies minimize risks such as contamination, mix-ups, and stability failures.
GMP regulations in both the US (21 CFR 210–211) and Europe (EudraLex Volume 4) require strict process validation, equipment qualification, and staff training to guarantee consistency.
Risk-based frameworks like ICH Q9 further strengthen safety by ensuring hazards are proactively identified and controlled.
Meeting legal and regulatory expectations
Compliance with GxP is legally mandatory worldwide. Regulators will not authorize the development, manufacture, or distribution of drugs without evidence of adherence.
As a result of tragedies such as the thalidomide crisis of the 1960s, GMP was formalized and expanded into the wider GxP family. Read about the origins of GMP in the United States.
Today, compliance is the license to operate. Without it, companies face warning letters, suspended licenses, and market bans.
Ensuring data integrity
Data is the backbone of pharmaceutical decision-making. If data is incomplete, manipulated, or unreliable, patient safety is at risk.
Regulators enforce strict data integrity principles, summarized under ALCOA+: data must be Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, and Available.
EU GMP Chapter 4, Annex 11, and FDA 21 CFR Part 11 all make it clear: whether data is on paper or electronic, it must be trustworthy and auditable. Most FDA warning letters today cite failures in data integrity, underscoring how central it is to compliance.
Reducing business and operational risks
Non-compliance is not only a regulatory problem, it’s a business threat. Inspections that uncover deficiencies can halt production, delay product launches, or even trigger recalls.
Financial costs are immediate, but the long-term damage to trust and reputation is often greater. Investors, partners, and patients lose confidence quickly, and it can take years to rebuild credibility.
By contrast, companies with strong GxP systems operate more efficiently, face fewer unexpected disruptions, and maintain competitive strength.
Enabling global market access and growth
GxP compliance is a gateway to global markets. Mature quality systems, reliable documentation, and inspection readiness accelerate approvals and open doors to new regions.
Regulators and distribution partners are more willing to collaborate with companies that demonstrate consistent reliability.
In other words, GxP does not just protect what you already have; it creates the foundation for long-term growth and competitiveness.
Having looked at the why, let’s turn to the who. Compliance only matters if there are authorities to enforce it, and in pharma, there are many.
Who regulates GxP in pharma?
GxP compliance in pharma is enforced by a global network of regulatory authorities, each with its own laws, inspection frameworks, and enforcement mechanisms.
While the underlying principles of patient safety and product quality are universal, the exact requirements differ from region to region.
For pharmaceutical companies operating internationally, understanding these nuances is critical to staying compliant across multiple jurisdictions.
United States – Food and Drug Administration (FDA)
In the US, the FDA is the primary authority overseeing GxP. Its regulations cover every stage of the product lifecycle:
- GMP under 21 CFR Parts 210–211 for manufacturing medicinal products and 21 CFR Part 820 for medical devices.
- GLP under 21 CFR Part 58 for non-clinical studies, and
- GCP under 21 CFR Parts 50, 56, and 312 for clinical trials.
- FDA 21 CFR Part 11 governs electronic records and signatures, ensuring that digital systems are validated and secure.
The FDA enforces these rules through inspections, Form 483 observations, warning letters, import alerts, and, in severe cases, consent decrees or product seizures.
Europe – European Medicines Agency (EMA) and Member States
In Europe, oversight is coordinated by the European Medicines Agency (EMA) and carried out by EU/EEA national competent authorities, such as the AIFA in Italy, FAMHP in Belgium, and BfArM in Germany.
The legal backbone is EudraLex Volume 4, which defines GMP requirements in Chapters 1–9 and supplements them with specialized Annexes (e.g. Annex 1 for sterile manufacturing, Annex 11 for computerized systems, Annex 15 for validation, and Annex 16 for batch certification).
GDP is covered under separate EU guidelines -2013/C 343/01 for finished medicinal products, and 2015/C 95/01 for active substances-, while Good Pharmacovigilance Practices (GVP) are laid out in modular EMA guidance (GVP Modules).
ICH - International Harmonization
The International Council for Harmonisation (ICH) works to align requirements across major regions, including the US, EU, Japan, and Canada. Although ICH guidelines such as ICH Q9 (Quality Risk Management) and ICH Q10 (Pharmaceutical Quality System) are not legally binding on their own, they are adopted into national laws, making them essential references for companies that operate globally.
Other global regulators
Beyond the US and EU, many other agencies enforce GxP standards. These include the PMDA in Japan, Health Canada, ANVISA in Brazil, NMPA in China, CDSCO in India, and the TGA in Australia. The World Health Organization (WHO) also issues GMP guidelines widely adopted in emerging markets.
While each regulator interprets GxP regulations in pharma slightly differently with their unique documentation, language, or procedural requirements, the principles are the same: safety, quality, and integrity.
While each regulator has unique documentation, language, or procedural requirements, most build upon the same international principles of GxP.
The GxP framework covers a wide range of practices, each designed to safeguard a different stage of the pharmaceutical lifecycle.
Let’s look at the most important ones.
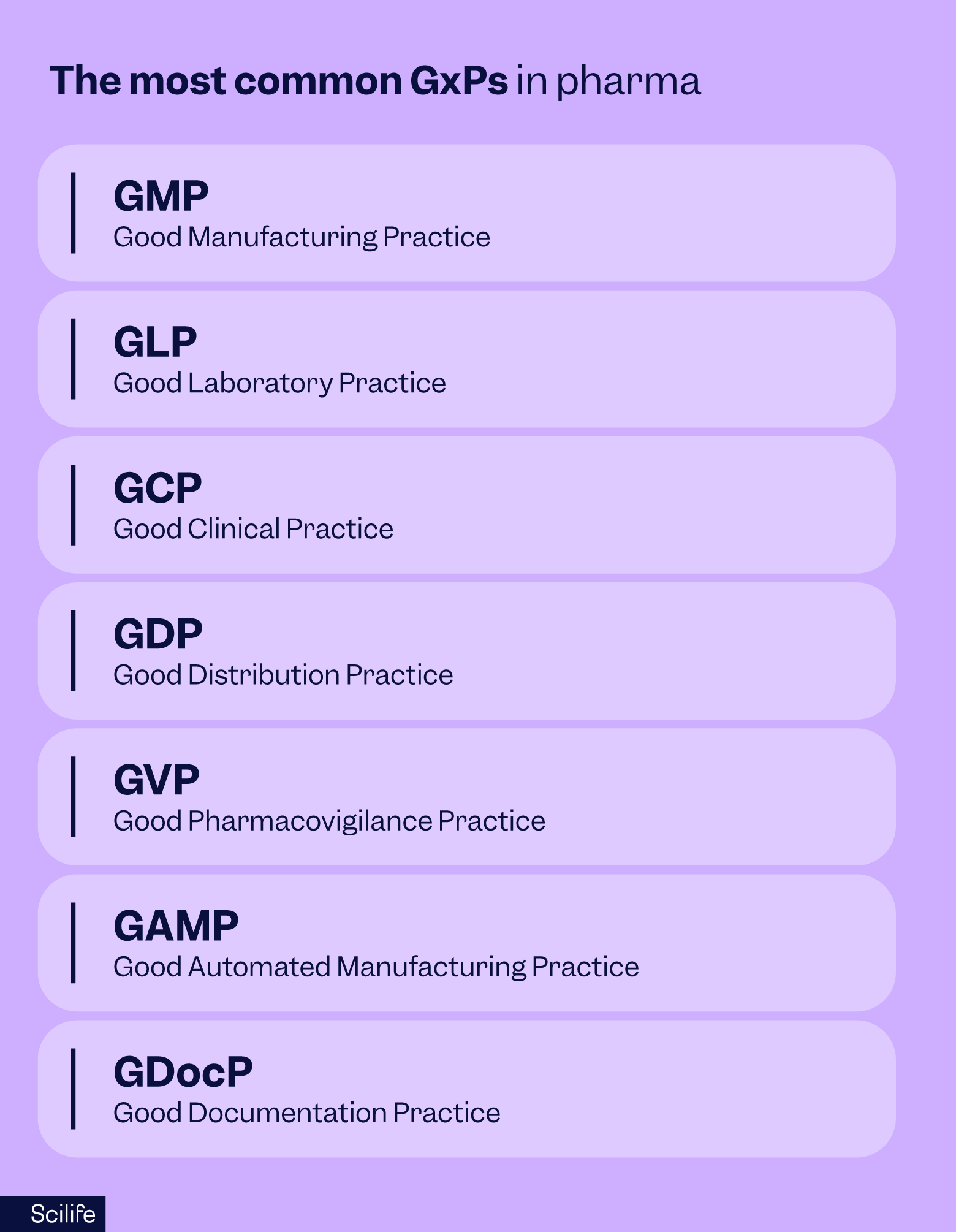
Types of GxP standards and regulations
GxP is not a single regulation but a family of quality guidelines and regulatory standards, each designed to safeguard a different stage of the pharmaceutical lifecycle.
From the research laboratory to the patient’s bedside, these frameworks ensure consistency, traceability, and accountability.
While every company will focus on the GxPs most relevant to its operations, regulators expect organizations to understand and apply the standards that affect their role in the supply chain.
Good Manufacturing Practice (GMP)
Before diving into its importance, let’s define what GMP is all about.
What is GMP?
GMP is the foundation of pharmaceutical quality. It governs how medicines are consistently manufactured and controlled, covering everything from facility design and equipment calibration to raw material handling, production processes, and quality control testing.
For a deeper breakdown, see our guide to the 5 main components of Good Manufacturing Practice (GMP), which explains the essential building blocks in detail.
Why is GMP important?
GMP ensures that every batch of medicine is produced to the same high standard, free from contamination or errors. It is the most heavily inspected and enforced part of GxP because it directly impacts patient safety.
To better understand how GMP requirements evolve with current practices, explore our article on the differences between GMP and cGMP.
Good Laboratory Practice (GLP)
What is GLP?
GLP applies to non-clinical laboratory studies such as toxicology, safety, and stability testing. It sets rules for how studies must be designed, performed, recorded, and reported to ensure that the data regulators rely on is trustworthy. In the US, GLP is codified in 21 CFR Part 58.
Why is GLP important?
GLP ensures that lab-generated data is reliable, reproducible, and traceable. If regulators can’t trust lab results, entire R&D programs can be delayed or rejected. For QA teams, GLP is the gatekeeper to safe human testing.
Good Clinical Practice (GCP)
What is GCP?
Once a drug candidate reaches human testing, GCP governs clinical trials involving human subjects. It sets international ethical and scientific standards to protect participants while ensuring trial data is accurate and credible. GCP draws on ICH E6 (R2) as its global benchmark, with FDA and EMA regulations enforcing specific requirements.
Why is GCP important?
GCP ensures patient rights and safety are prioritized in clinical research and that the data generated is acceptable for regulatory submissions. Without GCP compliance, clinical trial results cannot support drug approval.
To ensure the safety and well-being of participants in clinical trials, ICH E6(R2) introduces the following ethical and quality principles:
The thirteen principles are:
- Clinical trials should be conducted according to the ethical principles that have their origin in the Declaration of Helsinki.
- Studies can only be initiated if the potential benefits outweigh the risks.
- Safety, rights, and well-being of subjects are of paramount importance.
- The study must be preceded by adequate information.
- Clinical trials should be scientifically sound and described in a clear, detailed protocol.
- Trials should be conducted in compliance with the approved protocol.
- Medical care and decisions for trial subjects should be by qualified physicians or dentists.
- Individuals conducting trials should be qualified by education, training, and experience.
- Freely given informed consent should be obtained from all subjects prior to participation.
- All clinical trial information should be accurately recorded, handled, and stored.
- Confidentiality of records identifying subjects should be protected according to applicable regulations.
- Investigational products should be manufactured, handled, and stored according to Good Manufacturing Practice and used per the approved protocol.
- Systems and procedures should ensure the quality and integrity of clinical trial data.
Good Distribution Practice (GDP)
What is GDP?
Even after a product is manufactured, the work isn’t done. GDP ensures that medicines are stored, transported, and distributed in conditions that maintain their integrity at all times.
These rules protect against temperature excursions, mishandling, and falsified medicines entering the supply chain. In Europe, GDP is defined in 2013/C 343/01 for medicinal products for human use and 2015/C 95/01 for active substances.
Get to know the differences between GDP, GMP, and cGMP in our article, plus learn where they overlap and what their benefits are.
Why is GDP important?
Even perfectly manufactured medicinal products can be rendered contaminated, deteriorated, or unsafe if storage or transport conditions are compromised. GDP also protects patients by preventing falsified medicines from entering the legitimate supply chain.
Good Pharmacovigilance Practice (GVP)
What is GVP?
GVP provides the framework for post-marketing safety monitoring. The EMA’s modular GVP guidelines detail how companies must collect, evaluate, and act on adverse drug reactions and safety signals throughout a product’s lifecycle
Why is GVP important?
GVP ensures that once a drug or device is on the market, risks are continuously monitored, detected, assessed, and prevented from adverse effects. It is central to maintaining patient trust long after a product is approved.
It protects patient safety by identifying risks that may not have been obvious in controlled clinical trials, such as long-term side effects, rare adverse events, drug interactions, and medication errors.
It helps improve healthcare practices, inform regulatory decisions, and maintain public trust.
Good Automated Manufacturing Practice (GAMP)
When dealing with computerized systems in regulated environments, validation is key.
To guide this process, many organizations follow the complete GAMP 5 guide for GxP-compliant computerized systems, which outlines a risk-based framework for ensuring data integrity, product quality, and regulatory compliance.
What is GAMP?
GAMP is a globally recognized framework developed by the International Society for Pharmaceutical Engineering (ISPE) that provides comprehensive guidance for validating computerized systems used in regulated GxP environments such as pharmaceutical manufacturing.
It ensures these automated systems are fit for their intended use, compliant with regulatory requirements, and maintain product quality, patient safety, and data integrity.
The GAMP 5 framework emphasizes a risk-based, lifecycle approach to computerized system validation (CSV), which means validation efforts are scaled and focused based on system complexity and the potential impact on product quality and safety. It evolved into Computerized System Assurance (CSA), reflecting a more holistic and modern approach to system compliance that includes ongoing system assessment and assurance beyond initial validation.
Why is GAMP important?
Since modern pharma relies on automation and digital tools, GAMP (Good Automated Manufacturing Practice) is important because it provides a structured, risk-based framework that ensures computerized systems used in pharmaceutical and other regulated industries are validated, reliable, and compliant with regulatory requirements.
Good Documentation Practice (GDocP)
What is GDocP?
GDocP is a set of standards and principles that underpin all GxP guidelines. It ensures that every record, whether on paper or electronic, is accurate, legible, contemporaneous, and reliable.
The foundational principle behind GDocP is "If it isn’t documented, it didn’t happen," highlighting the critical role of proper documentation in regulatory compliance and quality assurance.
GDocP is essential for maintaining data integrity, which is globally benchmarked by the ALCOA+ principles.
To learn how to put this into action, explore our guide on documentation management best practices for life sciences organizations, which covers digital workflows, ALCOA+ principles, and regulatory expectations.
Why is GDocP important?
GDocP is important because it ensures that every action taken during the manufacturing process is consistently and accurately recorded, making it traceable and verifiable.
Inadequate or poor documentation is one of the most common causes of FDA Form 483 observations and regulatory inspections, as it can lead to questions about the reliability and integrity of product quality and safety.
Other specialized GxPs
Additional standards extend GxP principles into niche areas.
- Good Engineering Practice (GEP) covers facility and equipment design , construction, commissioning, operation, and maintenance of pharma facilities and equipment, ensuring systems are safe, compliant, fit for purpose, and aligned with regulatory expectations.
- Good Storage Practice (GSP) defines the correct conditions and procedures for the warehousing and storage of pharmaceutical products.
- Emerging practices, such as Good Review Practice (GRP) or Good Auditing Practice (GAP), represent the continuous expansion and refinement of GxP principles across the industry.
It’s also important to understand where the line is drawn. What falls under GxP, and what doesn’t.
GxP vs Non-GxP: What is gxp and non gxp in pharma?
Not every process in a pharmaceutical company falls under GxP regulations.
Understanding what is GxP and non GxP in pharma is essential for allocating resources correctly and ensuring audit readiness.
The difference between GxP and non-GxP in a pharmaceutical company lies primarily in the scope of regulation, oversight, and documentation requirements related to product quality, safety, and compliance.
GxP activities are those that have a direct impact on patient safety, product quality, or data integrity. These include:
- Manufacturing operations
- Laboratory studies
- Clinical trials
- Distribution processes
- Pharmacovigilance activities
- Documentation or computerized systems supporting these functions
For example, batch manufacturing records, laboratory notebooks, clinical trial case report forms, and validated quality management systems all require GxP oversight.
GxP compliance is mandatory in these areas to meet regulatory requirements and ensure the reliability of data submitted for product approval or patient use.
Non-GxP activities, by contrast, are business processes that do not directly influence the safety, quality, or efficacy of a regulated product. These typically include:
- Human resources, marketing
- Marketing
- Finance
- General administrative functions
For example, payroll systems, hiring, or office logistics systems do not require validation to the same standard as laboratory instruments or clinical data capture platforms.
However, the distinction is not always clear-cut. Some processes may sit in a grey zone. An area where understanding what is GxP and non GxP in pharma becomes especially important.
For example, IT infrastructure that supports both HR and laboratory systems may need controls to ensure it does not compromise regulated data.
Similarly, training systems may be considered non-GxP if they only manage general staff development, but they become GxP-critical when used to track SOP training or qualifications for GMP activities.
Discover how we can help you stay GMP compliant with a real-life use case!


What are the requirements for GxP compliance?
No matter which branch of GxP applies to your work, the GxP compliance requirements rest on the same foundation: reliable, traceable, and trustworthy quality systems.
Regardless of whether a company is working under GMP, GLP, GCP, or another GxP subset, there are common pillars that underpin compliance.
Key principles of GMP
Let’s take a closer look at GMP, since its principles serve as the foundations for compliance across pharmaceuticals.

Pharmaceutical Quality System (PQS)
Meeting international GxP regulations in pharma expectations means building a strong Pharmaceutical Quality System (PQS).
Regulators expect companies to adopt the principles of ICH Q10, which integrates quality into every stage of the product lifecycle. This includes leadership commitment, continuous improvement, and risk-based thinking.
Documentation and record-keeping
Documentation and records are the backbone of compliance. Following the ALCOA+ principles, records must be attributable, legible, contemporaneous, original, accurate, complete, consistent, enduring, and available. Whether data is paper-based or electronic, it must remain traceable and tamper-proof throughout its lifecycle.
Examples of GxP records in pharma include:
- Batch manufacturing records
- Standard Operating Procedures (SOPs) and training logs
- Laboratory notebooks and analytical data
- Clinical trial case report forms (CRFs)
- Equipment calibration and maintenance logs
- Audit trails from computerized systems
- CAPA reports and deviation records
- Change Control documentation
- Training records
Training and SOPs
Both FDA 21 CFR 211.25 and EU GMP Chapter 2 make it clear: every employee needs to be properly trained for their role and must know the current version of the SOPs that apply to their tasks. It’s not enough just to conduct training; it has to be well-documented, regularly refreshed, and importantly, its effectiveness needs to be assessed to make sure people really understand and apply what they’ve learned.
For new team members, foundational training on quality systems and GMP basics tailored to their job responsibilities is essential.
And ongoing training ensures that skills stay sharp and knowledge keeps pace with changes in procedures or regulations.
When training is done well, it empowers everyone to contribute confidently to robust compliance and product quality.
To see how this can be managed efficiently, explore our article on the importance of training automation in GxP environments, which explains how automated systems ensure compliance, reduce administrative burden, and keep teams always inspection-ready.
Validation and qualification
Any process, equipment, or computerized systems that can affect product quality, patient safety, or data integrity must be validated or qualified. GMP requires process validation and equipment qualification, while computerized systems must follow frameworks like GAMP 5 or newer CSA principles. Regulatory references include EU GMP Annex 11 and FDA 21 CFR Part 11.
Audit readiness
Audit readiness isn’t something you prepare for at the last minute. It has to be part of your daily routine. Compliance should be sustainable and ingrained in everything you do, not a reactive scramble when inspectors show up.
This means maintaining inspection readiness around the clock through regular internal audits, supplier evaluations, managing CAPAs effectively, and running mock inspections to stay sharp.
Regulators expect quick and reliable access to complete and accurate records during inspections, whether onsite or remote. To deliver on this, traceability and data integrity must be baked into your daily operations, not added as a last-minute addition when the pressure’s on.
Challenges in achieving and maintaining GxP regulations in pharma
Even with well-defined regulations and mature quality systems, GxP compliance in pharma remains a moving target. Here are the biggest challenges companies face today.
Evolving regulations
Regulatory frameworks are constantly updated to reflect scientific and technological progress. For example, the EU GMP Annex 1 revision on sterile products introduced stricter contamination control strategies, while updates to GAMP 5 2nd Edition redefined how companies validate computerized systems, moving from CSV to CSA.
Companies must continuously monitor and interpret regulatory changes to ensure their systems remain aligned.
A guide to EU GMP Annex 1 - expert compliance tips inside!

Complex global supply chains
Pharmaceutical supply chains are increasingly global and interconnected. A drug substance may be manufactured in one country, formulated in another, and distributed worldwide.
This complexity increases the risk of counterfeit products, mishandling, or quality degradation during transport. Regulators now expect companies not only to ensure their own compliance but also to verify the GxP compliance of every partner and supplier in their chain.
Data integrity issues
One of the most common causes of regulatory action is failure to maintain data integrity. FDA warning letters frequently cite issues such as incomplete batch records, backdated entries, missing audit trails, or insecure access controls.
In the digital age, ensuring that electronic data is accurate, tamper-proof, and audit-ready is one of the most critical and resource-intensive compliance challenges.
Human error and training gaps
Even with validated systems, human error remains a major risk factor. Inadequate training, outdated SOPs, or language barriers can all lead to non-compliance.
Regulators such as the FDA (21 CFR 211.25) and EMA (EU GMP Chapter 2) require companies to not only train staff but also prove that training is effective. Failing to embed a true culture of quality often results in repeated observations.
Legacy systems and resistance to digitalization
Many companies still rely on paper-based or hybrid systems that make it difficult to guarantee traceability, version control, and real-time oversight. These systems are increasingly viewed as non-viable by regulators, particularly when it comes to data integrity.
Transitioning to digital platforms can be resource-heavy, but failing to do so puts companies at risk of falling behind both regulatory expectations and industry standards.
Recommended learning:
The million-dollar mistake of skipping an eQMS - better to be safe than sorry

Risks of non-compliance
A violation of GxP risks much more than a simple warning. If regulators spot gaps, you may receive an FDA Form 483 observation, and later a warning letter. But if issues aren’t fixed quickly, things can escalate to import bans, product seizures, or even the loss of your manufacturing license.
In Europe, the consequences can be just as immediate: a Qualified Person (QP) may simply refuse to certify a batch. And if a batch isn’t certified, it doesn’t reach patients. It’s that simple.
But the impact doesn’t stop at regulatory sanctions. Think about the business side: product recalls, damaged reputation, shaken investor confidence, and closed doors to key markets. These setbacks can take years to recover from, if recovery is possible at all.
And in the worst-case scenario, if negligence is proven, individuals can face criminal charges — even imprisonment.
How technology supports GxP compliance in pharma
Trying to keep GxP compliance requirements under control with binders, spreadsheets, and scattered files is exhausting. It might get you through one inspection, but it’s stressful, error-prone, and not sustainable.
Regulators aren’t looking for good intentions. They want real-time visibility, data integrity, and audit trails you can trust. That’s where technology really makes your life easier.
This is where technology steps in to simplify and strengthen processes.
To see what to look for when adopting cloud-based tools, check out our guide on using SaaS solutions in GxP-compliant industries, which explains how to evaluate vendors, ensure data integrity, and stay audit-ready.
Digital validation management
Think about validation. Traditionally, it meant piles of paper, endless cross-checks, and weeks of preparation. With digital validation management, all of that gets simplified.
Instead of chasing signatures or flipping through binders, you can manage protocols, test scripts, and approvals in one place. Everything is traceable and audit-ready. And when the FDA or EMA asks for proof, you don’t sweat; you just click and show them.
Automated documentation & audit trails
Documentation is where most companies stumble. But manually updating logs or hunting down missing signatures is a nightmare. With an eQMS, version control is automatic, signatures are captured electronically, and audit trails are built in.
You don’t have to worry about whether people are working off the right SOP or whether data is missing, because the system keeps everything aligned.
That means inspections become less about firefighting and more about showing confidence in your process.
Platforms like Scilife simplify GxP compliance in pharma, making inspections faster, training seamless, and documentation always audit-ready.
Integrations with lab and manufacturing systems
Now imagine this scenario. Your lab results sit in the LIMS, your production data lives in the MES, and training records are in the QMS. If those systems don’t talk to each other, you end up spending hours stitching the story together for an auditor.
But when everything is integrated, you’ve got end-to-end traceability at your fingertips. You can follow a batch from raw material receipt to final release and prove every step was done correctly.
That kind of visibility not only makes audits smoother, it also builds trust inside your organization and with regulators.
How Scilife helps ensure GxP compliance in pharma
Meeting GMP requirements can be time-consuming and resource-heavy when managed manually.
That’s exactly why Scilife’s eQMS was built. Scilife was designed to simplify this process with regulated industries in mind. Here’s how it helps QA and RA teams manage compliance without chaos.
With Scilife’s eQMS, everything lives in one place. Your SOPs, CAPAs, changes, risks, and training records are all connected, traceable, and always up to date.
No more spreadsheets or guessing whether someone is working off the right version of a document. Every action leaves a full audit trail.
Most of the validation is already done for you. Scilife provides a risk-based validation package and documentation to significantly reduce your validation workload; you complete the process-specific steps.
The biggest win is peace of mind. Training, for example, runs itself. When an SOP is updated, Scilife automatically assigns it, sends reminders, and keeps records ready for inspection.
Dashboards give you a real-time view of where things stand. For instance, which CAPAs are open, which risks need attention, who is trained, and who isn’t.
So instead of scrambling at the last minute, you always know exactly where you stand.
Recommended learning:
Scilife use case examples & testimonials
Radboud Translational Medicine (RTM) - Dutch-based company that develops and manufactures GMP-complaint radiopharmaceuticals
Customers often say the same thing. Scilife’s eQMS is user-friendly, efficient, and saves significant time in managing quality processes. Audits suddenly feel manageable.
For instance, Radboud Translational Medicine appreciates features like automatic reminders, audit trails, version control, and easy document search, which reduce manual tasks and errors.
Managers value the ability to get clear overviews of training and document status through intuitive dashboards. Many note that Scilife not only helps maintain quality but also frees up time to focus on continuous improvement.
The customer support and onboarding process are frequently praised for being smooth and responsive, contributing to successful system adoption.
“Switching to Scilife made everything a lot easier, as now we have a clear overview and the system reminds people when to do things. Now, it’s one click instead of having to update new versions of every document.”
Suzanne Buijs, Head of QA
CTC (Clinical Trial Consultants): a full-service Clinical Research Organisation (CRO) based in Sweden
CTC, a growing clinical research organization, struggled with manual document and training management, which was time-consuming and centralized in QA.
After implementing Scilife’s eQMS, they gained automated document version control, easy remote access for staff and auditors, and simplified training management with automatic notifications.
This digital transformation reduced manual work and errors, improved audit readiness with remote document access, and gave managers clear visibility of their team's training status.
They went from spending weeks preparing for inspections to just a few hours because everything was already organized and accessible.
“The possibility of providing access to our QMS documents to auditors and inspectors has helped us a lot. And it has been very much appreciated by the auditors. They can access our QMS documents both before, during, and after the audit.”
Sofia Nyberg, QA specialist
Proteos Biotech: a leading biotech company that researches and produces recombinant enzymes
Another company that shared how they handled their QMS is Proteos Biotech.
They transitioned from a paper-based system to Scilife’s digital eQMS to improve document traceability, audit readiness, and overall quality management.
The implementation was smooth with excellent support and training. The user-friendly system, including its dashboard and document control features, has streamlined daily operations, making audits faster and less stressful.
The digital platform has enhanced collaboration across sites and made compliance much easier, satisfying both internal teams and external auditors.
With Scilife, you’re not just compliant, you’re confident. And that’s what every QA professional really wants when the inspector walks through the door.
“The dashboard allows us to see pending actions at a glance. It’s intuitive and helps us keep everything organized across all departments.”
Maria Vendrell Moreno, QA Manager
How do you achieve GxP compliance in the pharmaceutical industry?
Achieving GxP compliance requires more than understanding regulations. It means building quality into every stage of the product lifecycle through documented processes, trained personnel, risk-based decision-making, and continuous oversight.
Most pharmaceutical companies achieve and maintain GxP compliance by focusing on these core elements:
-
Documented procedures that define how critical activities are performed.
-
Employee training to ensure personnel are competent and follow approved processes.
-
Data integrity practices that keep records accurate, complete, and traceable.
-
Risk management to identify and mitigate quality and patient safety risks.
-
Validated computerized systems that support compliant, reliable processes.
-
Regular audits and continuous improvement to identify gaps before regulators do.
Rather than treating GxP as a checklist, successful organizations embed these principles into their daily operations, creating a culture where quality and compliance become part of every decision.
Conclusion
GxP regulations in pharma can sometimes feel like a never-ending struggle. The rules keep changing, inspectors always demand more, and teams spend countless hours chasing signatures or piecing together training logs. It’s easy to feel like you’re just trying to survive.
But GxP isn’t just about inspections, it’s about patients. Every log, every validation, every SOP you review is part of the safety net that ensures medicines are safe and trustworthy. That’s huge. And you’re right at the center of it.
The good news is, you don’t have to do it the hard way anymore. Digital tools, AI, and cloud platforms can transform compliance into a strength. With automation and integration, you gain control, resilience, and even a competitive edge.
Imagine walking into an inspection and knowing you’re already covered. That’s the confidence technology gives you.
And when you shift from firefighting to proactive compliance, everything changes.
You’re no longer addressing issues, you’re preventing them. That saves your team time, protects your company’s reputation, and actually makes compliance a competitive edge.
Regulators and partners notice when you’re in control, and they trust you more.
So, don’t think of GxP as a burden. With the right tools and mindset, compliance becomes your superpower.





