In the medical devices industry, “design transfer” refers to the process of transitioning a medical device from development to large-scale manufacturing.
For manufacturers, particularly personnel in the quality, R&D, and manufacturing functions, a working knowledge of design transfer and compliance with requirements is essential.
In this post, we’ll explain why design transfer is important, how to undertake design transfer effectively, and provide 5 best practices if you’re new to design transfer.
Key takeaways
- Design transfer is the process of translating a completed medical device design into production specifications, ensuring that devices validated during development can be manufactured consistently at scale.
- Under the FDA's 2026 Quality Management System Regulation (QMSR), design transfer requirements incorporate ISO 13485 by reference, placing greater emphasis on documented procedures and verification that designs are suitable for manufacturing.
- Successful design transfer requires cross-functional coordination between R&D, quality, regulatory, manufacturing, and supply chain teams to ensure design intent is accurately reflected in production.
- A digital Quality Management System (eQMS) helps manage design transfer documentation, ensuring version control, traceability, and audit-ready records across design and manufacturing activities.
What is design transfer?
In the medical devices industry, design transfer refers to a documented, structured, and verified process of translating a finished device design into a form that can be reliably manufactured at scale. There must be evidence that the production process is capable, and all relevant documentation must accurately reflect the approved device design. In simple terms, it is the point where engineering specifications become production specifications.
The goal of design transfer is to:
- Ensure the design intent is clearly communicated to manufacturing teams.
- Verify that the device can be consistently produced at scale.
- Demonstrate regulatory compliance and traceability through documented records.
While this concept sounds straightforward, design transfer is one of the most critical transitions in the product lifecycle. It connects two very different environments, with design and development on one side, and manufacturing on the other. When executed well, this process maintains quality, meaning that the device produced in the factory is identical to the one validated during development.
Scilife Academy: Product life cycle of medical devices


What changed in 2026? Design transfer under FDA QMSR (2026 update)
In February 2026, the FDA's Quality Management System Regulation (QMSR) came into effect in the United States, aligning US medical device quality requirements with ISO 13485. This update represents one of the most notable regulatory harmonization efforts in recent years.
One key implication of this shift is how regulators now define and structure design transfer. Under the legacy 21 CFR Part 820 Quality System Regulation, design transfer requirements were set forth in 21 CFR 820.30(h). Manufacturers were required to ensure that device design outputs were properly translated into production specifications.
Under the new QMSR framework, which incorporates ISO 13485 by reference, these expectations are aligned with ISO 13485 Clause 7.3.7 (Design and Development Transfer). This brief clause compels organizations to:
- Establish documented procedures for transferring design outputs to manufacturing.
- Verify that design outputs are suitable for manufacturing.
- Ensure production specifications accurately reflect the approved design outputs.
Although the regulatory language has changed, the core expectation remains the same: design transfer must be documented, controlled, and verifiable.
Terminology updates
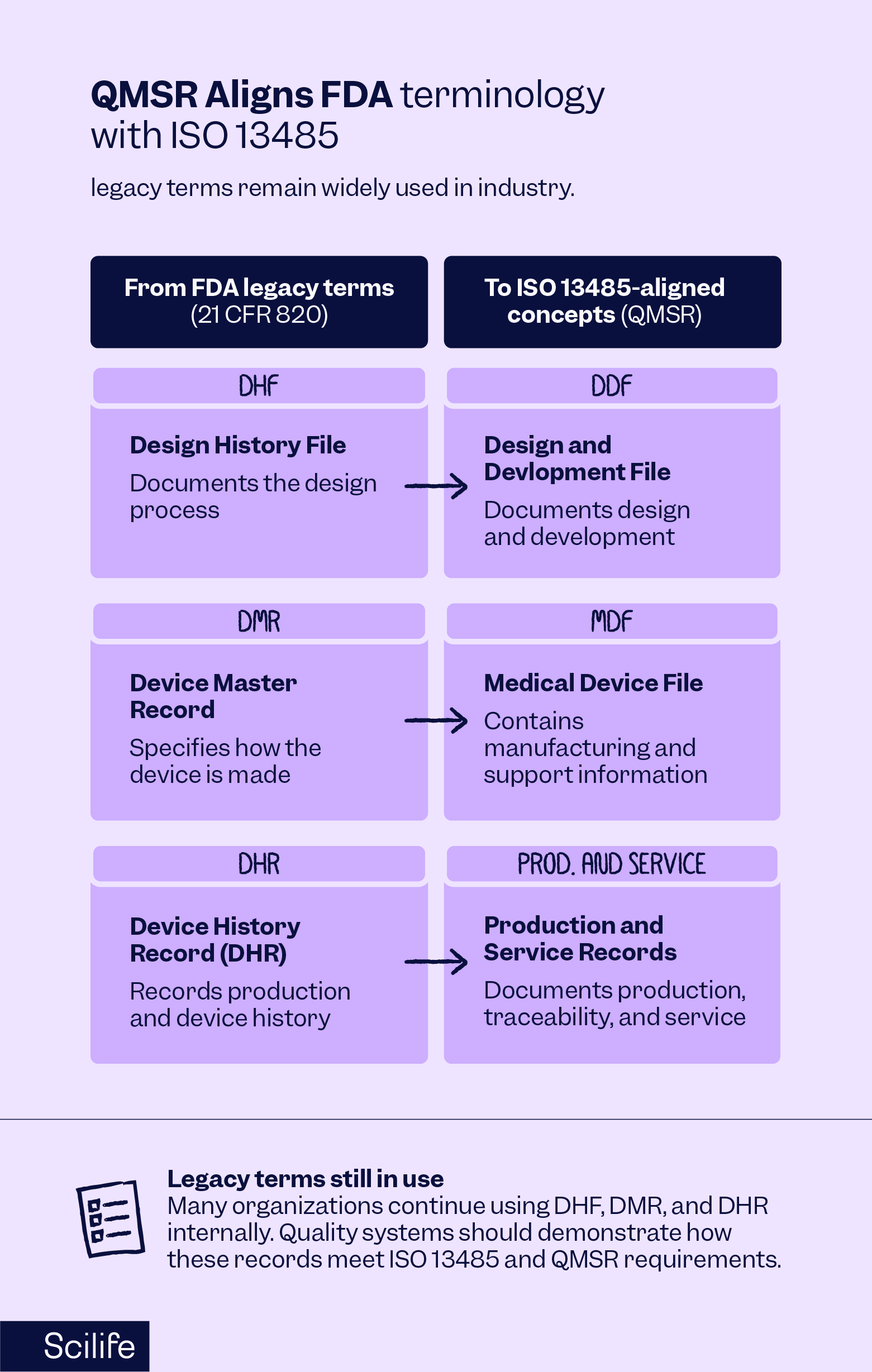
The terminology used in documentation has also evolved. Under the previous FDA framework, the regulations referred to:
- Design History File (DHF)
- Device Master Record (DMR)
- Device History Record (DHR)
QMSR shifts FDA terminology toward ISO 13485-aligned concepts such as Design and Development File (DDF) and Medical Device File (MDF), although legacy FDA terms like DHF, DMR, and DHR remain widely recognized and are still commonly used within industry quality systems. While many organizations continue using these legacy terms internally, their quality management systems should demonstrate how the associated records meet the applicable ISO 13485 and QMSR requirements.
ISO 13485 audit checklist


What does design transfer mean in practice under QMSR?
Design transfer as a process
In practice, design transfer is less about a single milestone and more about a structured series of activities that ensure development outputs are ready for manufacturing. One of the most common misunderstandings is assuming that design transfer begins once development is complete. In reality, many experts recommend planning for design transfer early in the product development process.
Design for manufacturability
For manufacturers, one practical impact of QMSR is an increased emphasis on verifying manufacturability before production specifications are finalized. In other words, regulators expect evidence that the device was developed according to Design for Manufacturability (DfM) principles, and can be produced consistently before full manufacturing begins.
Harmonized documentation structures
As mentioned, manufacturers that sell products across multiple markets, including the US, may find it easier to organize their files now that the FDA has aligned with international standards.
Cross-functional coordination
In many organizations, meeting the evidence requirements for design transfer involves coordination across multiple teams, including:
- R&D and engineering
- Quality assurance
- Regulatory affairs
- Manufacturing and operations
- Supply chain partners or contract manufacturers
Because these groups commonly operate in separate systems, maintaining traceability between design decisions and manufacturing specifications can be challenging. This is why many manufacturers rely on structured documentation workflows and centralized quality systems to manage design transfer documentation, ensuring all records remain traceable, version-controlled, and audit-ready.
Medical device design transfer checklist
Because regulatory and ISO guidance on design transfer is high-level, many organizations rely on internal checklists to verify that all necessary activities have been completed before manufacturing begins.
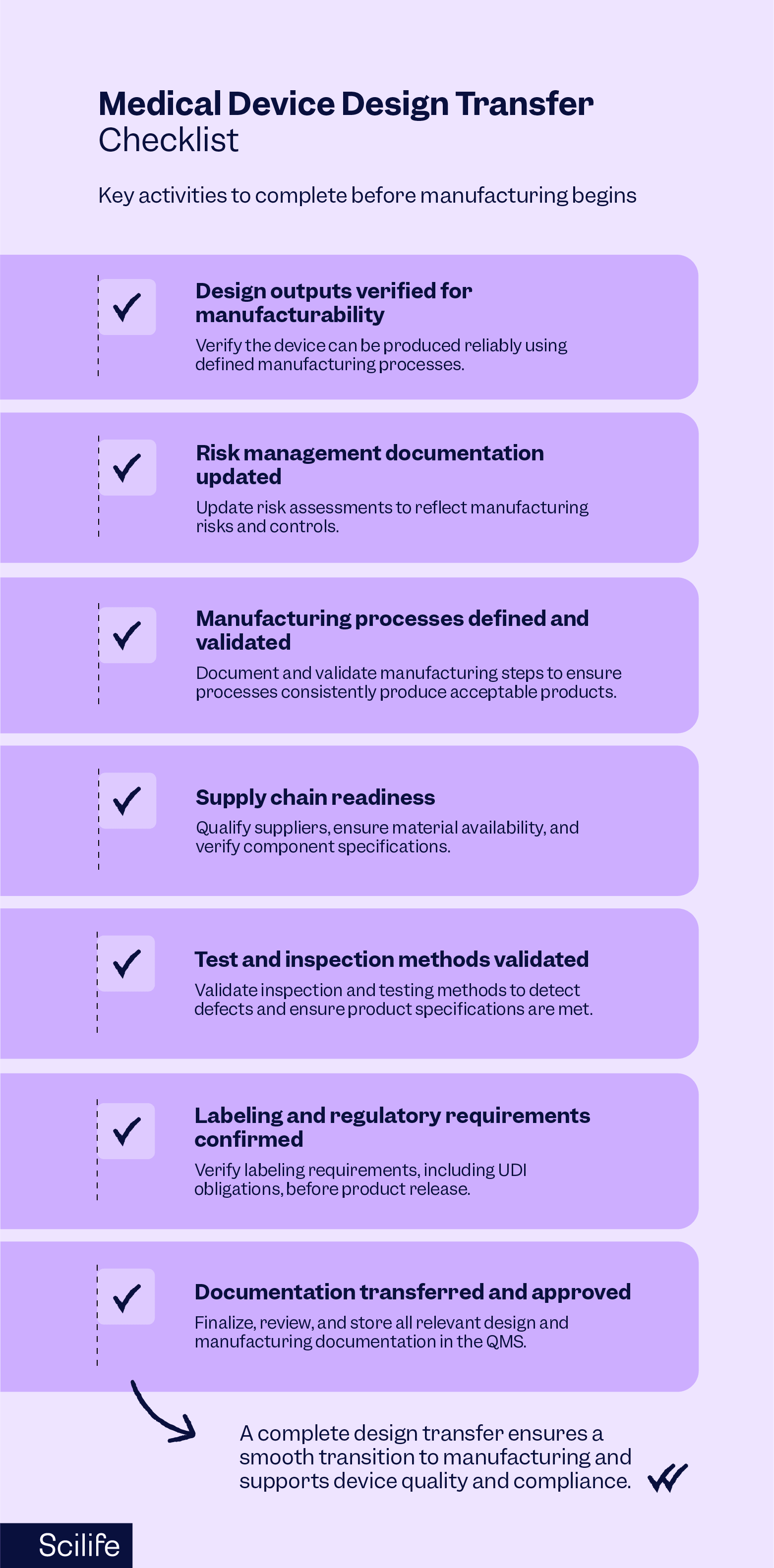
A typical medical device design transfer checklist may include the following elements:
- Design outputs verified for manufacturability: Before final production specifications are released, manufacturers must verify that the device can be produced reliably using defined manufacturing processes.
- Risk management documentation updated: Risk management does not stop at the design phase. Risk assessments should be updated to reflect manufacturing risks and controls associated with production processes.
- Manufacturing processes defined and validated: Manufacturing steps should be documented and validated, often through process validation activities such as verification that manufacturing equipment and systems are installed correctly, testing that the equipment operates correctly, and confirming that the manufacturing process consistently produces acceptable products.
- Supply chain readiness: Critical suppliers must be qualified, materials must be available, and component specifications must be verified to prevent delays or quality issues.
- Test and inspection methods validated: Inspection procedures and testing methods should be capable of detecting defects and ensuring product specifications are met.
- Labeling and regulatory requirements confirmed: Labeling requirements, including Unique Device Identification (UDI) obligations, must be verified before the product can be released.
- Documentation transferred and approved: All relevant design and manufacturing documentation should be finalized, reviewed, and stored within the organization’s quality management system.
Completing this checklist helps manufacturers demonstrate to regulatory bodies and other bodies (such as the International Organization for Standardization) that design transfer has been properly executed and that the device is ready for consistent manufacturing.
Best practices for effective design transfer
Without a structured design transfer process, even well-engineered devices can encounter major problems during commercialization, including delays, quality issues, or regulatory breaches/compliance issues. In my experience, several best practices consistently improve outcomes:
Involve manufacturing teams early
One of the most common causes of design transfer issues is insufficient manufacturing input during development. Involving manufacturing specialists early allows teams to identify potential production challenges before designs are finalized.
Design with manufacturability in mind
This is hugely important. DfM principles should be incorporated throughout development, as otherwise manufacturers risk delays to market and rework costs. If medical devices are developed from the start with manufacturability in mind, it means tolerances, materials, and assembly processes are more likely to be realistic in production environments.
Maintain clear documentation throughout development
Assembling documentation at the end of development can lead to missing records and inconsistencies. Maintaining documentation throughout the lifecycle simplifies the design transfer process and improves audit readiness.
Strengthen cross-functional collaboration
Effective design transfer depends on collaboration between engineering, quality, regulatory, and manufacturing teams. Regular design reviews and structured communication help maintain alignment.
Use centralized quality systems
Managing design documentation, approvals, and production specifications across multiple disconnected tools can increase the risk of incomplete or disjointed documentation, which can lead to compliance issues. Centralized digital systems help ensure the correct versions of documents are used and transferred to manufacturing.
Importing medical devices into the US: Essential steps and tips.

Conclusion: How an eQMS helps manage device documentation
As regulatory requirements evolve, managing documentation for design transfer has become increasingly complex. Manufacturers must demonstrate not only that designs meet regulatory requirements, but also that they can be reliably manufactured and fully traced through audit-ready documentation.
In practice, achieving this level of traceability using spreadsheets, shared drives, or email workflows can be difficult. Version control issues, fragmented records, and inconsistent approvals can quickly create compliance risks. This is where a digital Quality Management System (eQMS) can have a significant impact.
A modern eQMS enables medical device companies to manage the entire design transfer process in a structured, controlled environment. Instead of scattered documentation, organizations can maintain centralized records that support regulatory expectations and simplify audit preparation.
With the right system in place, teams can:
- Maintain secure, centralized documentation across design and manufacturing
- Control document creation, review, and approval workflows
- Track design changes and maintain full version traceability
- Ensure production specifications are aligned with approved design outputs
- Provide auditors with clear evidence of a controlled design transfer process
At Scilife, our Smart QMS platform helps medical device manufacturers manage documentation across the entire product lifecycle (from design and development through manufacturing and commercialization) helping teams maintain compliance, reduce risk, and streamline regulatory inspections.
Explore how Scilife’s Smart QMS supports seamless design and development processes.