
Artificial intelligence (AI) and machine learning (ML) are on the verge of transforming healthcare by creating new, vital insights from the vast amount of data gathered during healthcare delivery activities every day. AI/ML-based life science innovations have been booming in recent years, with many innovations related to the following applications:
- Early disease detection
- Improved accuracy of disease diagnosis
- Pattern recognition in human physiology
- Personalized diagnostics and therapeutics
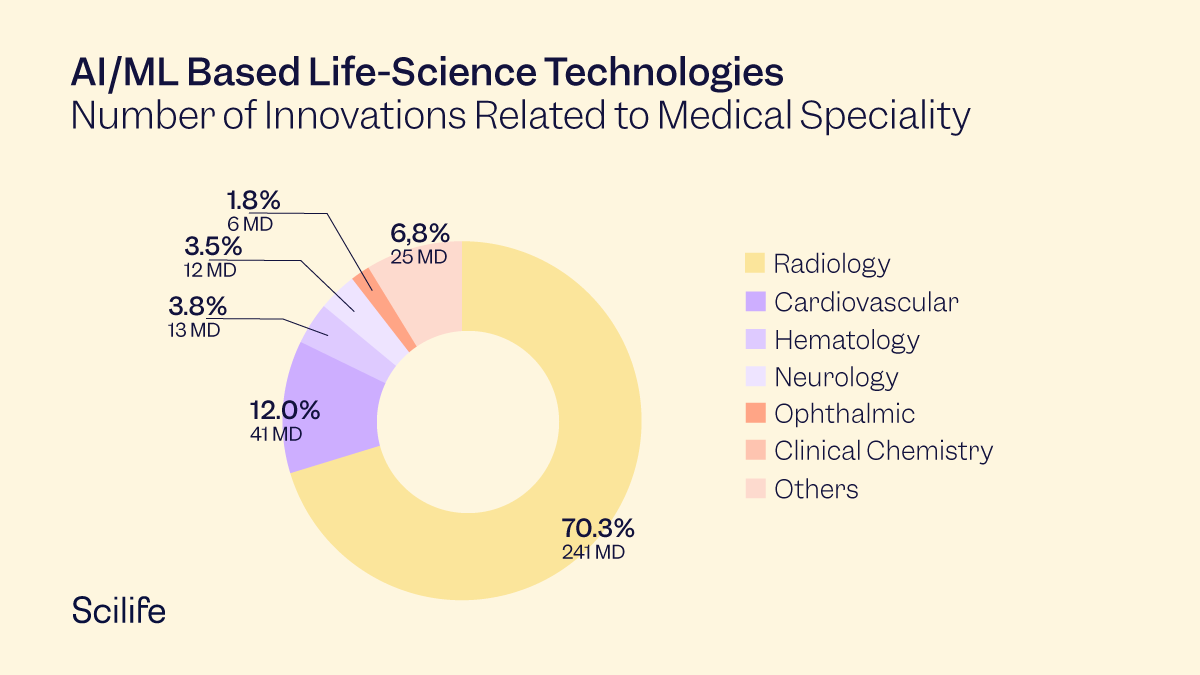
In 2020 and 2021, the largest number of innovations were in radiology and cardiology, in that order.
Yesterday's dream of digital healthcare technology has become today's reality. This has undoubtedly opened many new avenues in the field of life science innovations. The tsunami of clinical data has resulted in the development of Software as a Medical Device (SaMD). The big players in this space are GE, Philips, and Medtronics, which are already paving their way to breakthrough innovations.
Challenges Ahead
While every challenge comes with an opportunity, every opportunity also comes with a challenge. In AI/ML-based innovations in the life science industry, the challenge is self-evolving algorithms that are designed to learn from real-world applications. These algorithms are also known as "adaptive" or "continuously learning" algorithms which require auto-improvements from real-world experiences that the device captures throughout its use. There are already plenty of SaMDs that function with “locked” algorithms in use. However, when an algorithm is locked, it deprives its user of auto-improvements based on real-life data to make more accurate predictions. Therefore, AI/ML-based life-saving technology's real success is when it is not dependent on human intervention and learns from its own real-world experiences.
US FDA approved AI/ML Technologies
Until now, the FDA has only cleared or approved medical devices using locked algorithms, which provide repeatable results for the same input. Most of these technologies received approval through 510(k) premarket notification. However, according to a study published in the Journal of Medical Internet Research, "non-locked" learning algorithms are already used in self-diagnosis consumer apps today. However, the diagnostic efficiency of such apps seems to worsen over time. Therefore, systematic studies on a broader scale are necessary for healthcare providers and patients to correctly assess the safety, efficacy, and correct classification of such apps by healthcare regulating authorities. The FDA is aware of these developments and published a discussion paper to receive feedback on the proposed regulatory framework for modifications to AI/ML-based SaMD.
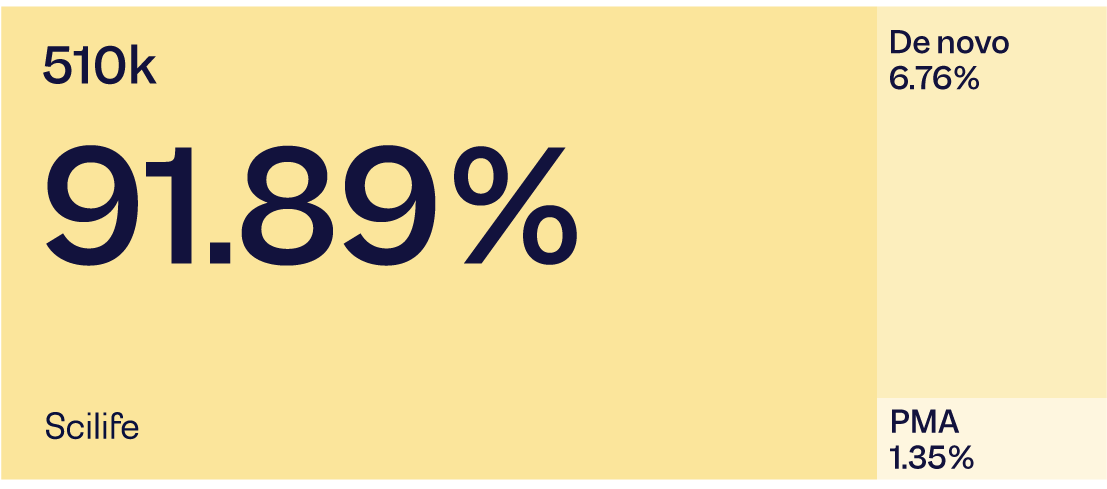
US FDA authorized technologies via 510 (k) clearance, granted De Novo request, or approved PMA
Note: Data from Jan 1, 2015, to March 31, 2020.
The FDA acknowledges that the highly iterative, autonomous, and adaptive nature of AI/ML-based tools requires a new, total product lifecycle (TPLC) regulatory approach. The agency envisions that this approach will facilitate a rapid product improvement cycle and allow devices to improve continually while providing adequate safeguards.
Proposed Regulatory Framework
Some of the critical aspects of the proposed regulatory framework are described in the following paragraphs:
Risk Based Categorization
Depending on the intended use, AI/ML-based SaMD can be classified into one of the four categories that reflect the clinical situation and device use risk. The risk increases from the lowest for Category I to the highest for Category IV. This classification system is based on the internationally harmonized International Medical Device Regulators Forum (IMDRF) risk categorization principles.
| State of Healthcare Situation or Condition | Significance of information Provided by SaMD to Healthcare Decision |
||
| Treat or Diagnose |
Drive Clinical Management | Inform Clinical Management | |
| Critical | IV | III | II |
| Serious | III | II | I |
| Non-serious | II | I | I |
Types of Modification
AI/ML-based SaMDs involve algorithm architecture modifications and re-training with new data sets, which would be subject to premarket review under the existing software modifications guidance. The types of modification may be classified into the following broad categories:
-
- Performance – clinical and analytical performance,
- Inputs to the algorithm and their clinical association to the SaMD output, and/or
- Intended use – The intended use of the SaMD.
The types of changes may not be mutually exclusive and may have varied impacts.
Total Product Life Cycle Approach
To handle these types of changes and their varied impacts, the FDA proposes "A Total Product Life Cycle Regulatory Approach'' (TPLC), through which it intends to evaluate the quality culture and organizational excellence of a particular company. The purpose is to have reasonable assurance of the high quality of their software development, testing, and performance monitoring of their products. This TPLC approach applies only to devices that require premarket submissions and enables the continuous evaluation and monitoring of a SaMD from its pre-market development to post-market performance, along with the continued demonstration of the organization's excellence.
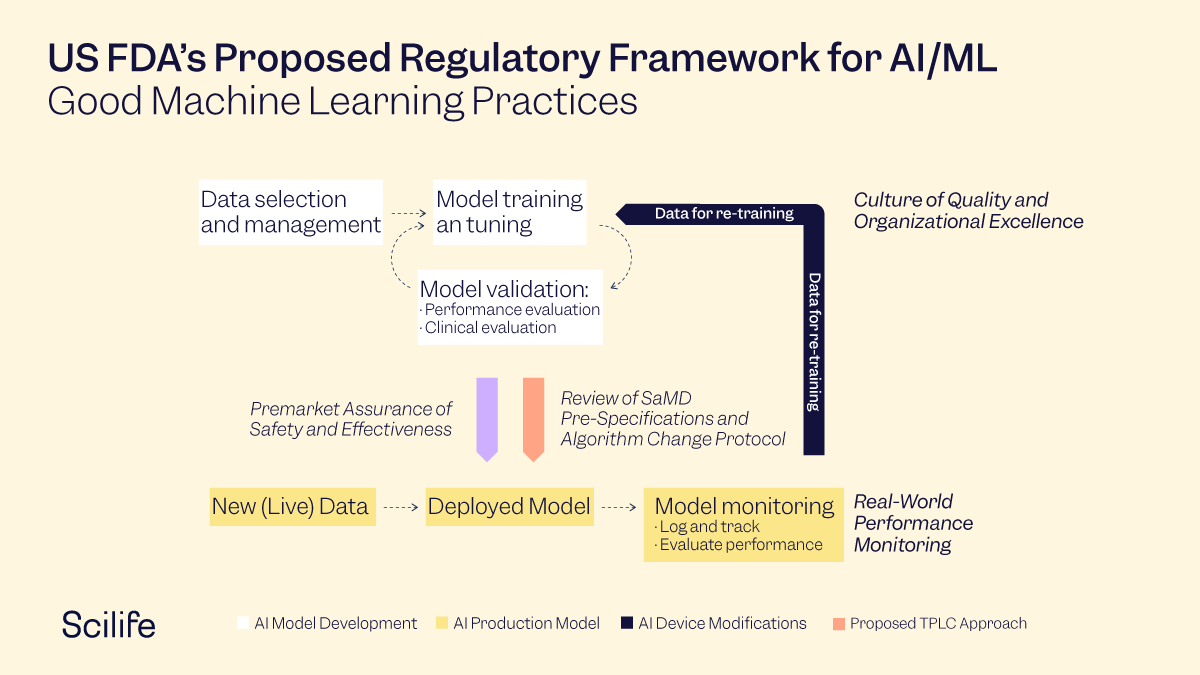
Overlay of the FDA TPLC Approach on AI/ML Workflow
General Principles for TPLC Approach
The FDA's proposed TPLC approach is based on the following principles to balance the benefits and risks for safe and effective AI/ML-based SaMD:
Establish clear expectations on quality systems and Good Machine Learning Practices (GMLP)
-
- All AI/ML-based SaMDs must demonstrate analytical and clinical validation as described in the SaMD Clinical Evaluation Guidance
Clinical Evaluation Valid Clinical Association Analytical Validation Clinical Validation Is there a valid clinical association between your SaMD output and your SaMD’s targeted clinical condition? Does your SaMD correctly process input data to generate accurate, reliable, and precise output data? Does the use of your SaMD’s accurate, reliable, and precise output data achieve your intended purpose in your target? IMDRF Description of Clinical Evaluation Components
- Some other considerations for GMLP may include:
- The relevance of data to the clinical problem and current clinical practices,
- The consistency of data acquisition, clinical relevance, and generalizable manner that aligns with the SaMD's intended use and modification plans,
- The appropriate separation between training, tuning, and test datasets, and
- An appropriate level of transparency of the output and the algorithm.
- All AI/ML-based SaMDs must demonstrate analytical and clinical validation as described in the SaMD Clinical Evaluation Guidance
Initial Premarket Assurance of Safety and Effectiveness
The proposed FDA framework for modifications to AI/ML-based SaMD relies on the principle of a "predetermined change control plan" that anticipates two types of modifications.
-
- SaMD Pre-Specifications (SPS) - can be defined as anticipated modifications to "performance" or "inputs" or "intended use" by the manufacturer of the SaMD. These are the types of modifications the manufacturer plans to achieve when the SaMD is in use. The SPS defines a "region of potential changes" around the original device's initial specifications and labeling. In other words, this is what the manufacturer plans the algorithm to become as it learns.
-
- Algorithm Change Protocol (ACP) – can be defined as the specific method that a manufacturer establishes to achieve and satisfactorily control the risks of the anticipated types of modifications defined in the SPS. In other words, the ACP is a step-by-step delineation of the data and procedures for the modification to achieve its goals and for the device to remain safe and effective after the modification. The table below provides an overview of the components of an ACP that describe how the algorithm will learn and change without compromising safety and effectiveness.
|
Data Management |
|
|
Re-training |
|
|
Performance Evaluation |
|
|
Update Procedure |
|
Algorithm Change Protocol Components
Manufacturers are expected to assess the modifications based on risk to patients, as mentioned in the software modifications guidance. The software modifications guidance expects a manufacturer to perform a risk assessment and ensure reasonable mitigation of risks.
Depending on the type of modification, the current software modifications guidance results in either:
1) Submission of a new 510(k) for premarket review, or
2) Documentation of the modification and the analysis in the risk management and 510(k) files.
In the case of AI/ML SaMD with approved SPS and ACP modifications, the manufacturers are expected to document the modification in their change history and all relevant records. These documents are required to be filed for reference to match with the "document" approach mentioned in the guidance for software modifications.
As per the software modifications guidance, if a modification is beyond the intended use for which the SaMD was previously authorized, the manufacturer is expected to submit a new premarket submission.
For this proposed approach, if SPS or ACP is refined based on the real-world learning and training for the same intended use of the AI/ML SaMD model, then FDA may do a "focused review" of the proposed SPS and ACP for that particular SaMD.
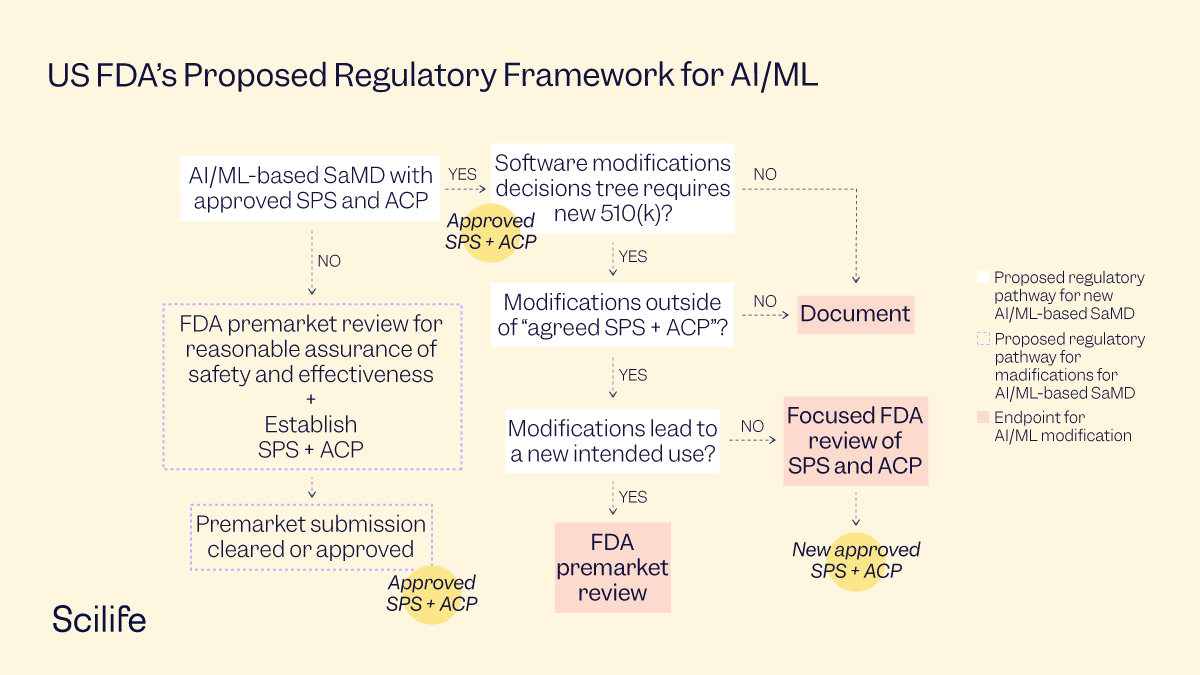
Approach to modifications to previously approved SaMD with SPS and ACP
Manufacturers may leverage the following options to engage with the FDA on the SPS and ACP for a particular SaMD:
- Contact the appropriate review division and obtain concurrence that the modification fits under current SPS and ACP,
- Start a pre-submission for a discussion on the modification and describe how it is within the SPS and ACP scope, or
- Go for a premarket submission or application of the modification to SPS and ACP.
Encourage increased transparency to users and the FDA using post-market real-world performance reporting to maintain continued assurance of safety and effectiveness.
To embrace the TPLC approach in the regulation of AI/ML-based SaMD, manufacturers are expected to support transparency and real-world performance monitoring. This is especially important for SaMDs that incorporate self-evolving AI/ML algorithms. Further, many of the modifications to AI/ML-based SaMD may be supported by the collection and monitoring of real-world data. Collecting performance data on the real-world use of the SaMD may enable manufacturers to understand the usability of their products, identify opportunities for improvements, and respond proactively to safety or usability concerns. This will also serve as an important mechanism for manufacturers to mitigate the modification associated risk for supporting the benefit-risk profile in the assessment of a particular AI/ML-based SaMD.
Conclusion
The draft guidance also elaborates on some practical case studies as examples to help manufacturers align on the approach. So far, stakeholders in the healthcare industry have broadly welcomed the proposed regulatory framework. AdvaMed has recommended the agency include Class III devices in the framework. The draft of the proposed framework has received a huge response in the form of 133 comments from corporates and individuals. However, despite the overall positive response to the regulatory groundwork, there was a small hassle in the process when the Department of Health and Human Services proposed that the FDA should cease to review some software tools altogether. This has created chaos, especially for manufacturers who had aligned their workflows with the draft guidance. The Department of Health and Human Services cited the cost of getting 510(k) clearance to justify the change, but it remains unclear if the Biden Administration will implement the proposed changes.
In any case, the coming few months will be hugely decisive in the AI/ML regulatory space for healthcare. We look forward to the agency's decision with the hope of a policy that will promote aggressive innovations in the life science industry without compromising quality or safety.


