

To successfully get a drug FDA approved, the data on the drug’s effects need to be reviewed by the Center for Drug Evaluation and Research (CDER), which in turn needs to determine that the drug’s benefits notably outweigh its known and potential risks for the intended patients. This approval process is performed within a structured framework.
What Is FDA Approval?
Have you seen the words “FDA approved” on a label or a company’s website? And are you sure that seal of FDA approval was actually real? The responsibility of the US Food and Drug Administration (FDA) is to protect public health by regulating human drugs and biological products, animal drugs, medical devices, tobacco products, food (including animal food), cosmetics, and electronic products that emit radiation. Only the most critical product groups should undergo premarket approval. Additionally, the FDA does not approve facilities, but does have the authority to inspect regulated facilities to verify that they comply with cGMPs.
Which Products Require FDA Approval?
The FDA has the authority to approve the following products:
- New human drugs
- Biological products such as therapeutic proteins, vaccines, allergenic products, cellular and gene therapies, and products manufactured from blood plasma
- Food additives intended for human consumption
- The FDA uses a risk-based, tiered approach for regulating medical devices for people
- The FDA also uses a risk-based approach for human cells and tissues
The FDA is not responsible for approving the following products:
- Compounded drugs
- Tabacco products
- Cosmetics
- Medical foods
- Infant formula
- Dietary supplements
- The food label, including the nutrition facts label
- Structure-function claims on dietary supplements and other foods
- Compounded drugs
New Human Drugs and Biological Products
All new human drugs and biological products are required to undergo premarket FDA approval. A new drug that does not comply with a monograph does require FDA approval. This means that the manufacturer should submit their data to the FDA to prove that the drug (or biological product) is safe and effective by conducting lab, animal, and human clinical testing for the drug’s intended use. Then, the FDA reviews the data and may approve the new product if the agency determines that the benefits of the product outweigh the risks for the intended use. Once approved, the company can manufacture the product according to the relevant quality standards.
In 2021, the Food and Drug Administration approved 50 novel drugs, and the number of approvals has increased significantly over the past decade.
Furthermore, the number of approvals over the past five years is higher than in the previous five years.
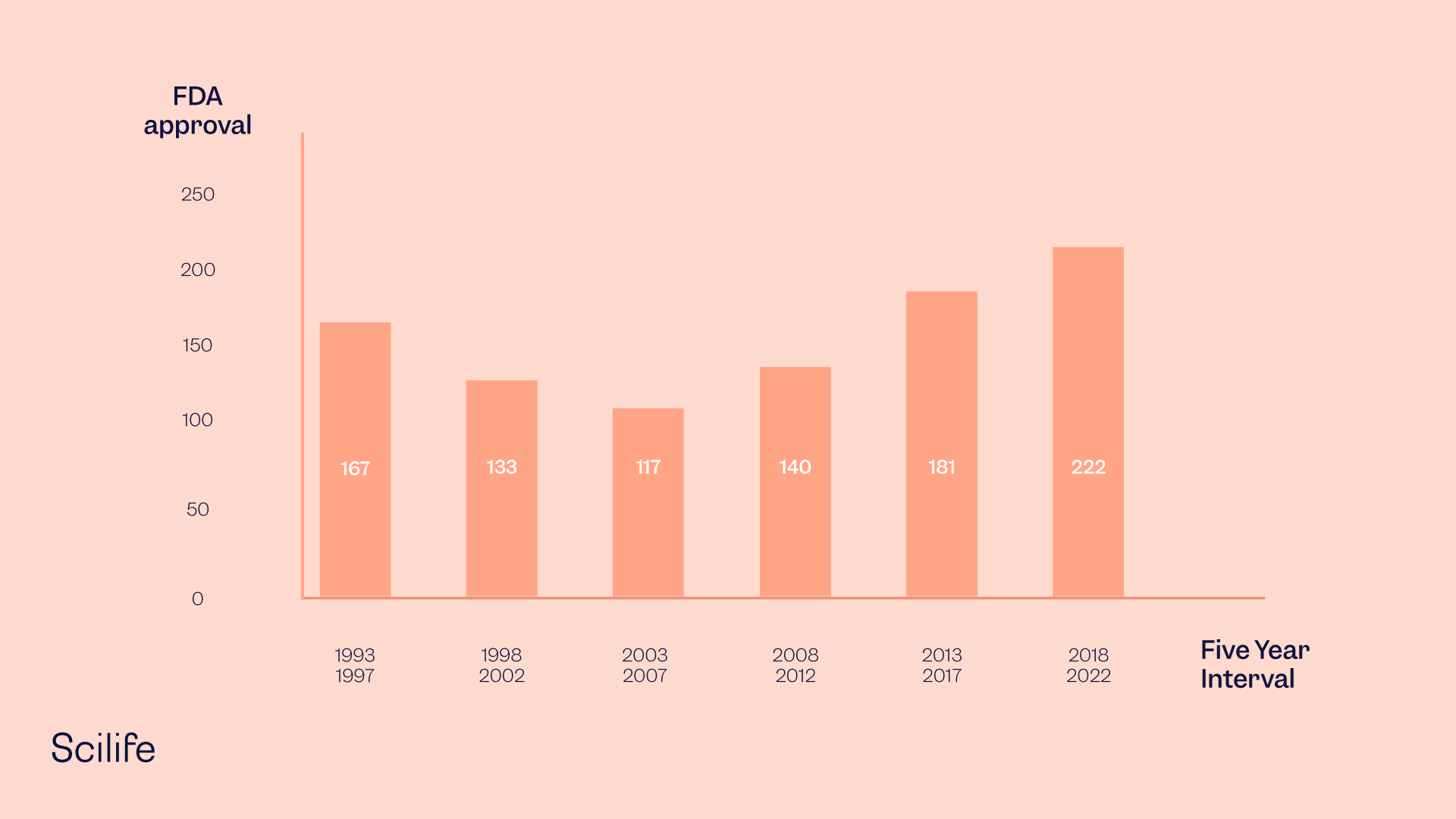
FDA Drug Approval Process
The CDER is the largest of the FDA’s six centers and oversees all prescription and over-the-counter drugs (OTC). Drug companies submit their applications to the CDER when they want to gain new drug approval. The CDER works with companies throughout the FDA drug approval process, from application to final drug approval. To achieve this, a team of CDER scientists, physicians, statisticians, chemists, and pharmacologists review the drug approval documents and propose labeling for pharmaceutical products.
What Are the Steps of the FDA Drug Approval Process?
A pharmaceutical company that needs FDA approval to market a new drug must complete the following five-step process:
- Discovery and Development
- Preclinical Research
- Clinical Research
- FDA Drug Review
- FDA Post Market Drug Safety Monitoring
If you want to dive into the details of this approval process, you can download our complete guide here.
 |
If you want to dive into the details of this approval process, you can download our complete guide here!
|

How Do You Expedite the FDA Approval Process?
The FDA can approve drugs faster than its European counterparts. The FDA standard review process requires 10 months for a standard review. However, the FDA also deploys the Prescription Drug User Fee Act (PDUFA) to expedite approval processing for drugs that target serious or life-threatening conditions.
Under the PDUFA, the FDA collects three types of user fees from new drug and biological product applications in order to facilitate faster approval:
- Application Fee – Clinical Data Required
- Application Fee – No Clinical Data Required
- Program Fee
Fast Track Programs include the following:
1. Fast Track Designation
This designation greenlights a priority review of drugs for unmet medical needs and serious conditions.
2. Accelerated Approval
This program allows patients with serious diseases more rapid access to promising therapies. The earlier approval of these therapies is based on a surrogate endpoint that is used when the clinical outcomes might take a long time to study.
3. Breakthrough Therapy Designation
This gives patients with serious or life-threatening diseases faster access. To receive this designation, preliminary clinical trials should offer substantial treatment advantages (i.e., safer or more effective) than existing options.
4. Priority Review Designation
The average time for drug to get within six months for the drugs under the priority review designation. To be part of this program, the drug should be highly effective at treating, diagnosing, or preventing a condition, also these drugs may work better with fewer side effects than drugs already on the market and treat a new population (such as children or the elderly).
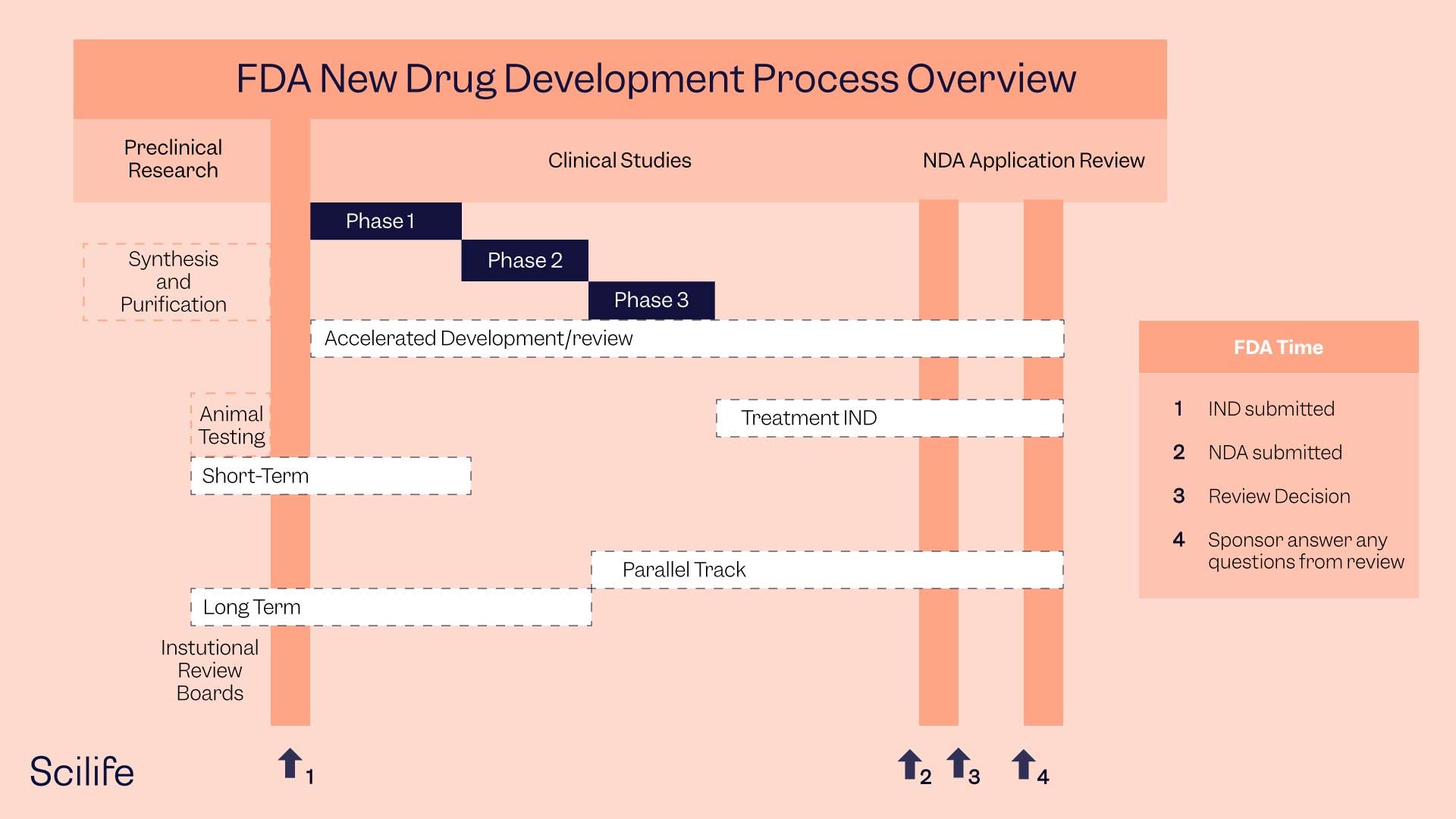
What Are the Different Types of FDA Drug Applications?
Have you heard of NDA or ANDA before? If you are somewhere within the FDA drug approval process, you knew the different types of FDA drug applications. Companies can submit different FDA drug applications according to the condition of the drug such as Investigational New Drug (IND), NDA, ANDA, and BLA. Let’s have a closer look at the FDA drug applications.
NDA: New Drug Application
An NDA is made by the sponsor of the new drug who obtains and provides enough evidence of the drug's safety and effectiveness to meet the FDA's requirements for marketing approval. The application should contain data from specific technical viewpoints for review, including chemistry, pharmacology, medical, biopharmaceutics, and statistics. Once the NDA is approved, the sponsor can sell the new drug in the US.
ANDA: Abbreviated New Drug Application
An ANDA is made by the sponsor of the abbreviated drug. The ANDA contains data for the review and approval of a generic drug product. Abbreviated New Drug is also known as “Generic drug” and applications are not required to include preclinical (animal) and clinical (human) data to establish safety and effectiveness since the innovator drug has proved its safety and effectiveness. However, the sponsor of the generic drug should scientifically demonstrate that its product is bioequivalent to the innovator drug, i.e., that the generic drug works in the same manner as the innovator drug. Once the ANDA is approved, the sponsor can manufacture and sell the generic drug in the US.
IND: Investigational New Drug
Before a drug can be transported or distributed across state lines, it must have an approved marketing application.
BLA: Biologic License Application
A BLA contains detailed information on the manufacturing processes, chemistry, pharmacology, clinical pharmacology, and the medical effects of the biological product. Once the BLA is approved, a license is issued allowing the sponsor to market the product.
A biological product can be marketed only if it has been approved for marketing under the Public Health Service Act.
New Drug Application (NDA)
The sponsor proposes the NDA application that includes all data gathered during the animal studies and human clinical trials of an IND to become part of the NDA.
The purpose of the NDA is to provide FDA reviewers with enough information to make the following key decisions:
- Comprehending whether the drug is safe and effective in its proposed use(s)
- Comprehending whether the benefits of the drug outweigh the risks
- Determining whether the drug's submitted labeling (package insert) is appropriate, and what it should include
- Determining whether the methods used in manufacturing the drug and the controls used to maintain its quality are sufficient to maintain the drug's identity, strength, quality, and purity.
Resources for NDA Submission
FDA drug approval documents for NDA submission should include the following data that provide the drug’s complete history:
- Clinical test results
- Drug ingredients
- Animal study results
- The behavior of the drug when it is applied
- Manufacturing, processing, and packaging processes
Furthermore, the following resources can guide you through the new drug application process, while assistance from CDER can help you meet the requirements, internal NDA review principles, policies, and procedures. Keep in mind, these documents are not regulations or laws; they are not enforceable, either through administrative actions or the courts.
FDA Regulations, Policies, and Procedures
Code of Federal Regulations (CFR)
The CFR contains 50 titles that represent broad areas that are subject to federal regulations.
21 CFR contains all regulations pertaining to food and drugs. The regulations document all actions of all drug sponsors that are required under 21 CFR Part 314 - Applications for FDA Approval to Market a New Drug or an Antibiotic Drug.
CDER's Manual of Policies and Procedures (MaPPs)
MaPPs documents are approved instructions for internal practices and procedures followed by CDER staff to help standardize the new drug review process and other activities and also define external activities. All MaPPs are open to the public to create a better understanding of office policies, definitions, staff responsibilities, and procedures.
The following MaPPs are specific for NDA applicants:
- Review of the Same Supplemental Change to More than One NDA or ANDA in More Than One Review Division
- NDAs and BLAs: Filing Review Issues
- Action Packages for NDAs and Efficacy Supplements
- Refusal to Accept Application for Filing From Applicants in Arrears
- Requesting and Accepting Non-Archivable Electronic Material for CDER Applications
Accelerating Compliance Time
Getting FDA approval for new pharmaceutical products requires careful recording of compliance of your development processes. Manually managing and tracking these activities takes time and creates opportunities for human error. This makes it obviously unsustainable, especially if you want to deliver your device to market on time.
Traceability accelerates the process, linking your development records and documents to tests and issues; you will be able to instantly track what has been implemented and tested, and if you meet all regulatory requirements.
But getting FDA approval is not the end. You will need to stay compliant as regulations — and your product — change.
Traceability can help you maintain FDA compliance in the long term, and it prepares you for future audits.
When that moment comes, you will need to be able to have all the documentation ready to prove you meet the requirements. By establishing traceability early on, you will make the audit as painless as it can be.
Plus, by tracking and linking requirements, code, tests, and issues, you can:
- Reduce recalls,
- Prevent lawsuits,
- And identify hazards before they become problems.
Scilife allows you to ensure inspection readiness at all times, as well as to control and centralize all your documentation and processes, ensuring traceability throughout the product lifecycle. Discover how Scilife Smart Quality Platform for Pharma can help you become fully compliant.
.png)

