

In my experience as a quality professional, I have found that a CAPA medical device system is the most revealing part of a company's internal structure. I once worked with a team that had nearly 40 open files, some dating back eighteen months, because they treated the process as a secondary administrative task rather than a priority.
During an audit, the inspector didn't care about our robust processes, they focused on those aging files to see if we were actually fixing our systemic failures or just documenting them. This is why understanding the CAPA medical device workflow is all-important for patient safety and regulatory compliance across the medical device, pharma, and biotech sectors.
The persistent challenge to achieve medical device companies CAPA process management best practices is that these investigations often move too slowly, creating operational bottlenecks and audit risks. When CAPAs remain open for an unreasonable amount of time, they signal to regulators that the quality management system is ineffective at identifying and resolving problems quickly.
In this post, we will explain the requirements for CAPA for medical devices, walk through the standard lifecycle, and outline 10 best practices to help you close investigations faster without compromising the integrity of your compliance data.
We will also explore how modern CAPA QMS integration tools medtech can automate these workflows to keep your team focused on resolution rather than paperwork.
Key takeaways
What is CAPA in medical devices?
Corrective and Preventive Action is a systematic framework used to collect information, analyze quality data, and eliminate the root causes of nonconformities to prevent their recurrence. Within the context of a medical device CAPA, the process is governed by strict standards such as ISO 13485:2016 and FDA 21 CFR Part 820.100.
ISO 13485 specifically separates the concept into two clauses: 8.5.2 for Corrective Action, which addresses existing failures, and 8.5.3 for Preventive Action, which focuses on mitigating potential risks before they occur. This system is highly scrutinized during inspections because it serves as the primary indicator of whether a manufacturer can proactively manage its own quality failures.
A detailed review of historical enforcement data reveals exactly where most companies fall short during inspections. An analysis of FDA warning letters from 2013 to 2019 shows that 72% of all CAPA violations were due to a failure to establish and keep basic procedures under 21 CFR 820.100(a).
Documentation of activities and results accounted for another 10.6% of the citations, while failures in the initial data analysis phase made up 8.4%. These statistics suggest that the majority of regulatory risk is found at the very base of the system, underscoring the need for a process that is both well-defined and meticulously recorded.
Create an Effective CAPA form


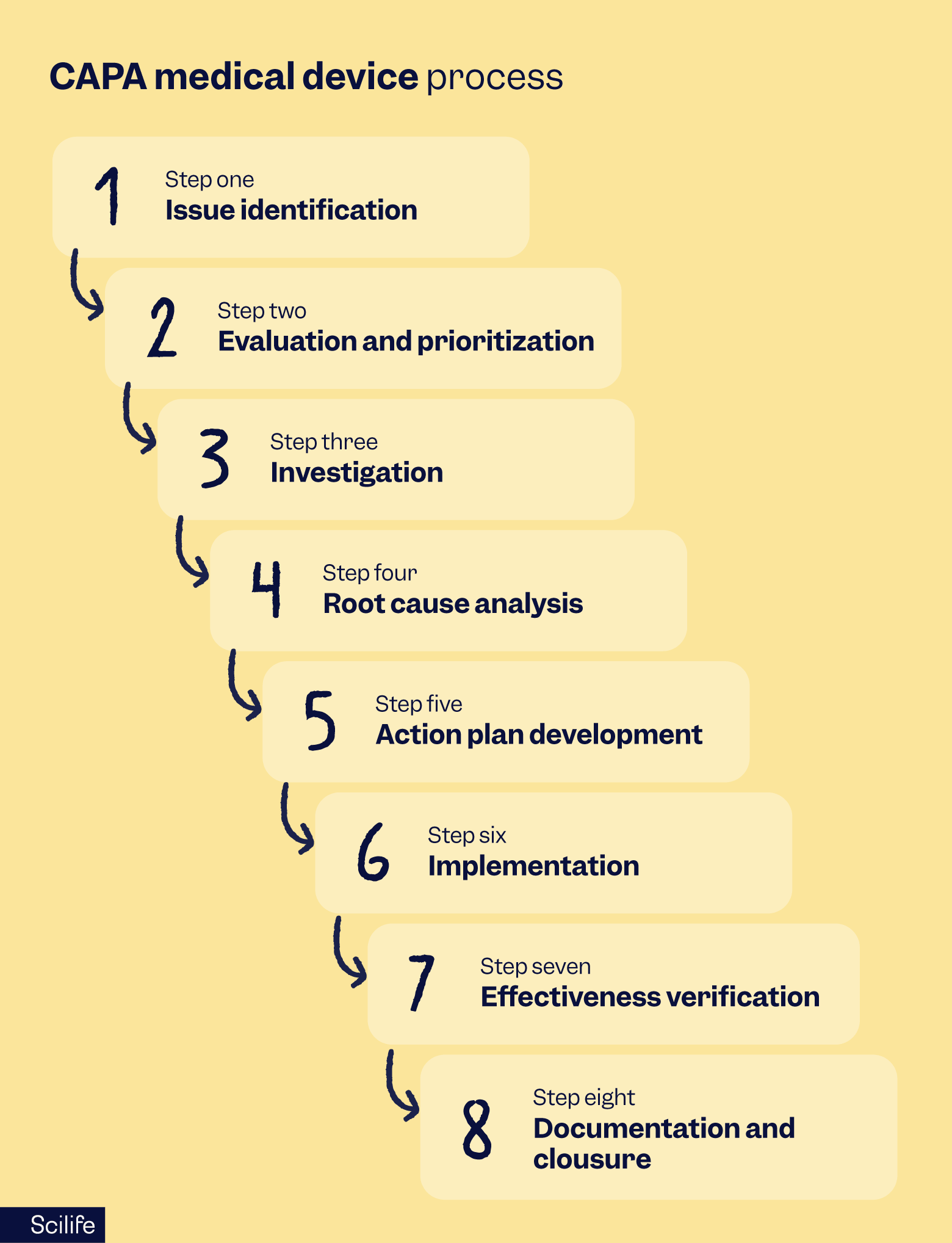
CAPA medical device process
The typical lifecycle of a medical device CAPA process moves through several distinct phases to verify that a problem is fully extinguished at its source. Most delays occur during the root cause analysis or the effectiveness check, where teams often struggle to gather sufficient objective evidence.
-
Issue Identification: Capturing and logging a potential problem from sources like complaints, audit findings, or nonconforming product reports.
-
Evaluation and Prioritization: Assessing the risk to human health and business continuity to determine if a formal CAPA is warranted or if the issue can be managed via another quality process.
-
Investigation: Documenting a plan that specifies the procedures, personnel, and resources required to study the failure.
-
Root Cause Analysis: Using structured tools to move beyond the symptoms and find the underlying process or systemic failure.
-
Action Plan Development: Creating a detailed map of required changes to procedures, training, or manufacturing processes.
-
Implementation: Executing the plan and recording all changes to verify that the root cause is being addressed.
-
Effectiveness Verification: Confirming through data that the actions worked and did not adversely affect the finished device.
-
Documentation and Closure: Compiling all findings into a final report for management review and formally closing the file.
Examples of CAPA in medical devices
Practical applications of a CAPA medical device investigation often involve complex technical failures that span multiple departments. For example, if a manufacturer of implantable cardiac devices discovers battery defects, the CAPA investigation would likely lead to enhanced supplier audits and stricter process validation protocols.
Another common scenario is a manufacturing failure where an automated syringe assembly line produces defective plungers due to misaligned machinery. Here, the corrective action would involve recalibrating equipment while the preventive action might include updating the preventive maintenance schedules and implementing real-time quality monitoring.
These examples show that the process must be holistic, looking at both the immediate fix and the long-term systemic safeguard.
Top 10 CAPA best practices
Use a risk-based filter for initiation
Not every deviation requires a full CAPA. Overusing the system leads to backlogs that bury critical safety issues under a mountain of paperwork. When you establish clear risk-based criteria, you can make sure that your resources are dedicated to the systemic issues that truly impact patient safety.
Establish a cross-functional Management Review Board
A common CAPA mistake is allowing the quality department to own CAPA. A weekly meeting with representatives from engineering, manufacturing, and regulatory affairs helps find faster resolutions because those closest to the process are the ones defining the fix.
Standardize your Root Cause Analysis tools
Whether your team uses the 5 Whys, Fishbone Diagrams, or Fault Tree Analysis, the key is consistency. Standardizing these tools helps investigators move quickly and provides a predictable format for auditors to follow when they review your files.
Avoid the human error trap
Citing human error as a root cause is a red flag for regulators because it often means the investigation didn't go deep enough. Most problems blamed on people are actually caused by poor SOPs, inadequate training, or a high-stress environment that leads to slips and lapses.
Recommended learning:

Set realistic and flexible deadlines
Arbitrary deadlines are a recipe for non-compliance. Use data from previous investigations to set timeframes that are tight enough to keep momentum but realistic enough to be met without cutting corners on the investigation.
Use predictive data analytics
Moving from reactive to proactive management requires looking at leading indicators. Modern research shows that companies using data analytics to flag supplier instability or defect trends can reduce critical defect rates by up to 18% within a single year.
Verify effectiveness before you close the file
You must prove that the corrective action worked before you consider the job done. This involves setting specific requirements for what success looks like and collecting objective evidence, such as a period of zero recurring defects, to validate the fix.
Integrate CAPA with other QMS processes
Your CAPA system should pull data directly from complaints, nonconformances, and internal audits. This integration makes sure that you have a single source of truth and that no critical quality signals are missed.
Prevent expensive errors and recalls by managing CAPAs proactively


Train personnel on the why of CAPA
Operators and engineers often see CAPA as a pain. Training them on how these actions directly protect the patient and the company's market standing, you then build a culture of quality where people are motivated to participate in the solution.
Digitize your workflow
Manual spreadsheets and paper files are the biggest contributors to CAPA aging. Using digital tools allows for automated reminders, real-time tracking, and instant visibility for management, which accelerates the path to closure.

Digital systems: How software helps close CAPAs faster
As we move through 2026, the regulatory environment is becoming more demanding, with a massive focus on the harmonization of standards. According to the FDA, the move toward the Quality Management System Regulation (QMSR) is expected to result in a net social benefit of roughly $3.6 billion over the next decade by reducing duplicative paperwork and streamlining global reviews. However, handling this transition requires a level of data integrity that manual systems simply cannot provide.
Scilife provides the digital support needed to move through these complexities with confidence. Our platform automates the most time-consuming parts of the CAPA medical device lifecycle, from sending automated reminders to the right stakeholders to providing real-time visibility through KPI dashboards.

![CAPA: FDA's #1 Most Cited Inspection Finding [Full Guide]](https://www.scilife.io/hubfs/The-ultimate-corrective-and-preventive-action-%28CAPA%29-guide-every-life-science-professional-needs.png)



