

Corrective and Preventive Action (CAPA) is an essential component of quality management in the pharmaceutical and medical device industry. However, problems with the CAPA process can lead to regulatory action, product recalls, and, most importantly, loss of reputation.The CAPA management process is often further complicated by the need to meet regulatory requirements and quality standards such as ISO 9001 and ISO 13485.
While ISO 9001 sets broad quality management expectations across industries, ISO 13485 establishes more specialized requirements for medical device companies. For life sciences organizations, CAPA compliance with both standards can be demanding, as each calls for rigorous CAPA quality processes that must be carefully implemented and maintained.
I’ve heard plenty of stories from QA professionals about how frustrating and ineffective CAPA systems can be. If you’re about to implement a new CAPA quality management system —or are thinking about improving the one you already have—it's worth understanding the common pitfalls before you dive in.
Knowing where things typically go wrong can help you avoid minor issues snowballing into bigger failures.
In this blog, we’ll walk through some of the most common challenges with CAPA management and share practical tips to overcome them, so you can build a CAPA system that actually works.
If your CAPA process still produces repeat deviations, it may be time to revisit the fundamentals. This CAPA guide outlines a structured, evidence-driven approach designed to stop issues from resurfacing.
Key CAPA compliance takeaways
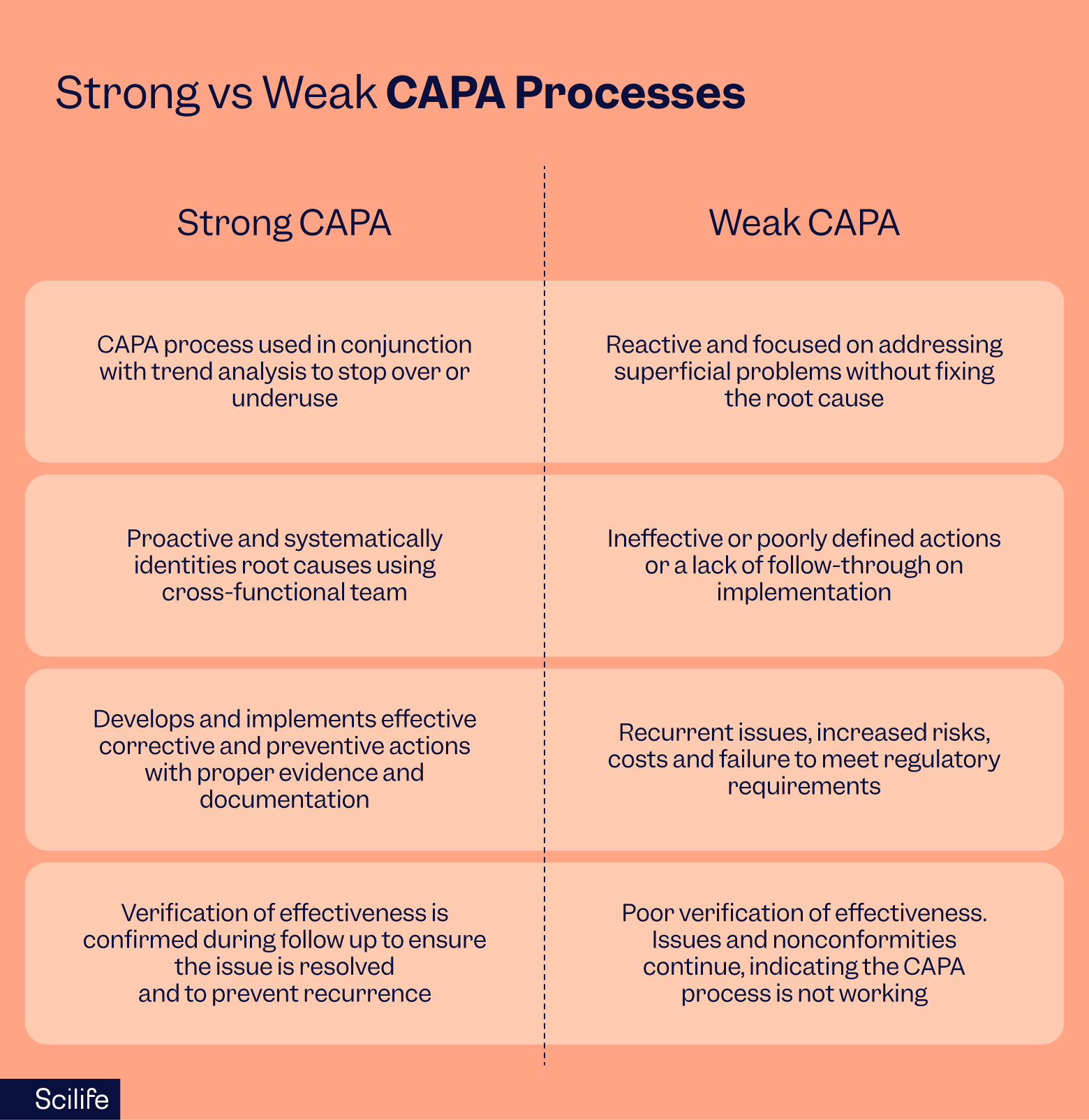
Strong CAPA process vs weak CAPA quality process
A strong CAPA process identifies the root causes of quality issues, implements effective corrective and preventive actions, and then verifies their success, leading to continuous improvement in quality and the manufacturing process, enhanced compliance with regulations and standards, and reduced operating costs.
In contrast, a weak CAPA quality management process may only address symptoms rather than the root causes of issues, leading to recurring problems, increased risk of product recalls, reputational damage to the organization, and higher operational expenses.
Common CAPA process mistakes
1. Inadequate Root Cause Analysis
The mistake:
In a recent online survey, we asked more than 300 QA professionals in the life sciences industry to share their biggest issues with CAPA. And the most commonly cited reason? Poor root cause analysis.

This is not particularly encouraging, as root cause analysis is the cornerstone of the CAPA quality process.
When QA teams jump straight to fixing the symptom rather than digging into the real reasons behind the issue, they can miss the actual problem.
Both ISO 9001 (Clause 10.2) and ISO 13485 (Clauses 8.5.2 & 8.5.3) are clear that you need to investigate the actual cause of nonconformities, not just patch them up, so that the same problem doesn’t resurface.
In practice, I’ve noticed that when people skip proper root cause analysis, they often end up firefighting the same issues repeatedly, further sinking in the murky waters of CAPA compliance mishaps.
How to avoid:
- Clearly define a precise, measurable, and documented problem statement based on data and evidence for better CAPA quality management.
- Form a cross-functional team to help you dig deeper into root causes, as everyone brings a unique knowledge base and skillset to the table.
- Collect evidence by gathering all relevant data and information, such as incident reports, witness statements, process records, and system documentation.
- All relevant stakeholders and departments should be present for CAPA root cause analyses.
- Apply structured methodologies like the '5 Whys', Fishbone diagrams, or Fault Tree analysis.
- Validate the root cause by examining the evidence to ensure the identified cause aligns with the data collected.
Example:
Identifying that a batch of pills failed quality testing due to contamination, tracing the contamination to a faulty air filtration system, rather than just attributing it to general "poor operator practices”.
This leads to a corrective action (replacing the filter) and a preventive action (implementing a new preventive maintenance schedule for the filtration system) to stop future occurrences.
Free Root Cause Analysis template


2. Reactive instead of proactive CAPAs
The CAPA mistake:
Organizations fail to perform proper root cause analyses before executing corrective actions, leading to recurrent issues.
Rather than taking the time to investigate and proactively prevent said CAPA compliance issues from happening again, the team is stuck in reactive mode, constantly fighting fires without ever cutting them off at the source.
How to avoid:
- If you’re seeing recurrent issues, look more deeply at your past root cause analyses. Have they identified the real root causes of the issues?
- Identify why the real root cause hasn't been fixed: is it because the root cause hasn't been identified? Is it due to a pragmatic issue, such as a lack of resources or funding to replace a piece of faulty equipment? Is it due to inefficient manual CAPA processes being organized across spreadsheets?
- Ensure staff are adequately trained in root cause analysis and hands-on applications.
Example:
A medicine batch fails quality control testing. During a root cause analysis, the team finds that a particular machine was slightly misaligned, and it gets adjusted. Unfortunately, in reality, parts of the machine need to be replaced as they are worn and come loose regularly, creating similar defects on a regular basis. The team is constantly busy with various recurring issues that never get fully solved.
3. Insufficient follow-up (Verification of Effectiveness)
The CAPA mistake:
One of the biggest pitfalls is declaring a CAPA quality process closed without following up on whether the fix actually worked. Based on our recent survey, this was the second most significant issue experienced after poor root cause analysis.
Unfortunately, quality management expectations set out in ISO 9001 (Clause 10.2) and ISO 13485 (Clauses 8.5.2 & 8.5.3) emphasize the verification of corrective and preventive actions' effectiveness and the documentation of the results.
How to avoid:
- Establish an effectiveness verification plan to determine whether the implemented actions were effective
- Define your effectiveness criteria, using clear metrics and targets to ascertain success
- Gather objective evidence by collecting data that shows the problem has been fixed or its frequency reduced after the corrective actions were taken.
- Don’t close the CAPA if the actions weren't practical; open a new CAPA instead, referencing the original, and work on finding a solution that solves the root causes.
Example:
A CAPA is initiated to address recurring contamination events during the production of a sterile drug. The production line is cleaned, and a temporary change in the cleaning procedure is implemented. The operator is retrained on proper aseptic techniques.
The CAPA compliance team marks the CAPA as closed because these immediate actions have been taken and documented. The company does not gather data to confirm that the contamination incidents have stopped and that the new cleaning procedures or operator training have successfully prevented their recurrence.
The recurring contamination issue persists, leading to potential product recalls, regulatory scrutiny (e.g., FDA 483 warning letters), and damage to the company's reputation, as the root cause of the problem remains unresolved.
Recommended learning:

4. Lack of CAPA process effectiveness
The CAPA mistake:
One of the most common problems with CAPA processes is the development of ineffective CAPA action plans. Scilife’s recent survey of QA professionals found that the third most common CAPA-related issue found during FDA inspections was a lack of CAPA effectiveness.
Action plans should be clear and concise, with specific details on the problem, the corrective action, the person responsible, and the timeline. However, many organizations fail to provide enough detail, leading to confusion and ineffective corrective actions.
How to avoid ineffective action plans:
Ensure that your CAPA plan is comprehensive, clear, and concise, and targets the root cause of the problem.
- To improve CAPA action plan development, involve cross-functional teams and use SMART goals.
- Use a risk-based approach to prioritize actions and ensure they are achievable within the required timelines to avoid unrealistic timelines.
- Assign action owner(s) so that the CAPA action plan is clear on responsibilities.
- Write all details about the actions and inform all related functions that might be affected by the change.
- Track the progress of your CAPA plan against your SMART goals to ensure that all actions taken are effective and prevent the issue’s recurrence.
Example:
A specific batch of a pharmaceutical product fails quality testing due to packaging defects. A shallow root cause analysis identifies that the human machine operator made a mistake during the packaging process.
As a corrective action, the operator receives additional training on proper packaging procedures. The CAPA plan concludes with no specific preventive actions beyond the retraining. Unfortunately, the real root cause was faulty packaging equipment, which led to recurring defects in subsequent batches.
5. Lack of cross-functional teams
The CAPA mistake:
Root cause analyses are conducted by the QA team alone, and other departments can at times decline to take part.
In my experience, this can not only lead to ineffective root cause analyses but also a feeling of frustration among QA teams. One respondent in the Scilife 2025 survey of QA professionals recently quipped, "If QA had a dollar for every time we ran a CAPA alone, we'd host a conference called 'Root Cause, Not Lone Cause.”
How to avoid:
- Foster a quality culture across departments, and recognize and reward employees who contribute to quality improvements and actively participate in the CAPA process
- Ensure your CAPA investigation team is fully trained and up-to-date on best practices in root cause analysis techniques such as building ‘Fishbone Diagrams,’ ‘Is-Is Not’ logic matrices, and the ‘5 Whys’ exercise.
- Involve relevant department representatives such as Quality, Production, Engineering, and Regulatory Affairs.
- Define clear roles and responsibilities within the cross-functional team to ensure accountability.
- Maintain a centralized repository in an accessible location for standardized documentation and workflows to document the entire CAPA process.
Example:
QA oversees the CAPA quality process, ensuring compliance with regulations. Manufacturing and Operations investigate the production process for the affected batch, identifying potential points of failure in the packaging line or equipment.
Engineering assesses and troubleshoots the machinery involved in the packaging process, determining if equipment malfunction or design flaws contributed to the defect.
6. Poor documentation
The CAPA mistake:
Another common issue is inaccurate or incomplete records that hinder the assessment of CAPA plan effectiveness and trigger regulatory action.
Full, properly organized documentation is your lifeline in an audit. ISO 9001 (Clause 10.2) and ISO 13485 (Clauses 8.5.2 & 8.5.3) expect you to maintain proper records of both the investigation and the actions you took.
I’ve seen cases where teams did the right things but couldn’t prove it later, which caused unnecessary stress during inspections. Using purpose-built software and treating CAPA documentation as part of the process rather than a bolt-on at the end can really help.
How to avoid:
- Check your CAPA documentation process using a few closed CAPA reports, ensuring you have recorded everything.
- If you are using a robust document management system with standardized templates for CAPA documentation, ensure all team members are trained to complete these templates wholly and accurately.
- Finally, to ensure your document management system remains effective, you should regularly review and update it.
Example:
A batch of a drug failed a customer's quality test, showing high impurity levels that were not present at the manufacturer's facility. The manufacturer and customer both used the same High Performance Liquid Chromatography (HPLC) method, whereas the customer prepared their samples at room temperature, while the manufacturer used ice.
This temperature difference was identified as the root cause. Unfortunately, there was a lack of scientific evidence to suggest that the temperature difference was the primary factor contributing to the impurity results. There were insufficient effectiveness checks and no follow-up to confirm that the corrective action was effective.
7. Poor or complex CAPA management processes
The CAPA mistake:
CAPA processes are often managed inefficiently by organizations without a central tracking system. Many companies rely on spreadsheets, and some still use paper.
How to avoid:
- Develop a CAPA management system that meets all requirements while keeping it as simple as possible. In this way, it will be possible for team members to follow the CAPA process in an appropriate manner.
- Understand that managing the CAPA process manually using spreadsheets can be challenging to track, as a single CAPA plan may include tens of actions, action owners, and different timelines.
- To boost efficiency, consider a tailored software solution such as a centralized CAPA management system.
Example:
Multiple departments use separate spreadsheets, leading to inconsistent data and silos of information instead of a single source of truth. Employees spend excessive time filling out forms, maintaining complex formulas, and sending countless emails to coordinate, rather than focusing on the CAPA work itself. During an audit, auditors may struggle to find the definitive, complete version of the CAPA file, leading to lengthy requests for information and potential compliance issues.
8. Delayed CAPA implementation
The CAPA mistake:
Even the best CAPA plan fails if it isn’t acted on quickly. Both ISO 9001 and ISO 13485 stress timely implementation (Clause 10.2 for ISO 9001; Clauses 8.5.2 & 8.5.3 for ISO 13485). In my experience, delays usually come from unclear responsibilities, resource bottlenecks, and disagreements on the best approach.
How to avoid:
- Ensure CAPA quality process responsibilities are clearly defined and training is up to date among personnel.
- Use a structured approach, like SOPs and formal training logs, to ensure CAPA plans are implemented efficiently.
- Be realistic about how much time and what people are needed for CAPA responsibilities, and tackle understaffing or resource bottlenecks.
Example:
A pharmaceutical company discovers that batches of its product are frequently contaminated. Through a CAPA process, the root cause is identified as inadequate sanitization protocols.
However, the implementation is delayed because the company lacks the necessary equipment and personnel for the automated cleaning validation. Additionally, different departments disagree on the specific details of the revised cleaning protocols.
The initial plan underestimated the complexity of retraining the entire production staff and installing the new automated system. The recurring product contamination continues for several more months because the actions to fix it and prevent recurrence were not completed on time, potentially leading to further product recalls or regulatory issues.
No more CAPA complexity! Become a pro at CAPA implementation with Scilife


9. CAPA underuse
The CAPA mistake:
The underuse of CAPA means that systemic issues and quality failures go unaddressed, creating a risk of product failure, harm to patients, and regulatory consequences. This can happen when personnel don’t know their responsibilities or due to human error.
How to avoid:
- Establish clear, specific triggers using objective criteria for what constitutes a CAPA, focusing on the quantity and severity of an issue.
- Prevent recurrence by conducting a thorough root cause analysis and fixing the underlying problem.
Example:
A medical device company repeatedly receives customer complaints about a faulty device feature, but does not initiate a formal CAPA investigation. They only implement minor, temporary fixes, failing to address the underlying design or manufacturing flaw. Repeat incidents of device failure and potential patient harm occur, leading to increased regulatory scrutiny, warnings, and potential recalls.
10. CAPA overuse
The CAPA mistake:
On the other side of the coin, you have CAPA overuse. A company opens a CAPA for every customer complaint or minor process deviation, resulting in wasted resources. Administrative work piles up, and key, high-risk issues get insufficient attention. We get CAPA is exciting (at least for quality people), but hold your horses.
How to avoid:
All of the criteria in the CAPA underuse category apply here, plus:
- Don’t start initiating a CAPA for every deviation. Instead, perform risk-based trend analysis to identify recurring problems that indicate a systemic issue rather than a one-off anomaly.
- As part of risk-based trend analysis, implement a “pre-CAPA” monitoring window for borderline issues, to identify whether similar events are occurring and determine if a full CAPA process is genuinely needed.
- Realize that minor, one-time issues can be resolved with simple corrections, and don’t always need a CAPA.
Example:
A company is "CAPA-happy," opening a CAPA for every single minor customer complaint or process deviation. The quality team is overwhelmed with hundreds of open CAPAs, hindering their ability to conduct proper root cause analysis on truly significant issues. This "death by CAPA" can lead to delayed resolution of serious problems and inefficient use of resources.
11. Lack of communication
The CAPA mistake:
The underuse of CAPA means that systemic issues and quality failures go unaddressed, creating a risk of product failure, harm to patients, and regulatory consequences. This can happen when personnel don’t know their responsibilities or due to human error.
How to avoid:
- Implement a robust communication plan that includes all team members and stakeholders.
- Regularly communicate updates on investigations, CAPA plans, and progress against timelines.
- Ensure everyone understands their roles and responsibilities and can continually provide feedback to improve the CAPA process.
Example:
A single batch of a medication fails quality testing because the packaging was improperly sealed. QC doesn't effectively communicate the severity or specific nature of the defect to the packaging team or manufacturing. Packaging engineering isn't involved in the investigation, so they aren't aware of the specific failure modes. Manufacturing receives vague instructions, leading to inconsistent implementation of the CAPA plan.
Expert tips: How do you improve a CAPA process?
If you are ready to take your CAPA processes to the next level, consider:
- Set SMART goals for improvement, and set key performance indicators (KPIs) to track your progress.
- To improve CAPA effectiveness, focus on objectives and metrics such as the CAPA effectiveness Index, the preventive vs. corrective ratio, problem recurrence rates, and the percentage of CAPAs closed by their due date.
- To improve CAPA efficiency, focus on objectives and metrics that measure timeliness, such as closure rates, the number of overdue CAPAs, and the average closure time.
- To improve CAPA productivity, focus on volume, such as the number of open CAPAs or the number of CAPAs processed in a certain unit of time.
- To improve CAPA compliance, focus on improving internal audits to ensure adherence to regulations in the case of regulatory audits.
- Standardize and streamline your CAPA processes by leveraging digital tools such as software solutions with automated workflows and centralized repositories built in, such as Scilife’s eQMS.
Conclusion: How CAPA compliance software can help you
CAPA mistakes are all too common in the life sciences industry, but they don’t have to be roadblocks. With the right approach, you can prevent potential regulatory action and protect your company’s reputation. Focus on identifying and addressing the true root cause, documenting your findings thoroughly, implementing a practical and effective action plan, and keeping communication clear across teams and stakeholders.
This is also where technology can make a real difference to your CAPA quality management system.
A digital solution like Scilife QMS software streamlines the entire CAPA process, automating documentation, reducing the risk of human error, and ensuring full traceability. By providing a centralized platform for collaboration and oversight, Scilife helps teams not only improve compliance but also increase the efficiency and effectiveness of their CAPA systems.
In short, pairing best practices with the right digital tools allows you to transform CAPA management from a regulatory obligation into a driver of continuous quality improvement.



![CAPA: FDA's #1 Most Cited Inspection Finding [Full Guide]](https://www.scilife.io/hubfs/The-ultimate-corrective-and-preventive-action-%28CAPA%29-guide-every-life-science-professional-needs.png)

