The In Vitro Diagnostic Regulation (IVDR) has been the law in the EU since 26 May 2022. By now, every manufacturer placing IVD devices on the European market should have a compliant quality management system in place. Yet in 2026, our experience shows that compliance gaps remain, particularly around data integration with EUDAMED and documentation of AI use in critical processes. These gaps are no longer abstract. They expose manufacturers to enforcement action, vigilance reporting delays, and market access risk.
The regulatory landscape has shifted again. On 16 December 2025, the European Commission published a targeted simplification proposal that signals what regulators want from QMS: agility, integration with digital systems like EUDAMED, and transparency about how AI is used in critical documentation decisions.
This guide is for quality and regulatory professionals who need to close compliance gaps or strengthen existing QMS implementations. We cover the five pillars of IVDR-compliant QMS, the practical realities of EUDAMED integration, and concrete steps to prepare for upcoming regulatory changes. Everything here is grounded in current regulatory requirements, not speculation about what might change.
Regulatory Requirements for IVDs in EU
Regulation (EU) 2024/1860: Now in force
Regulation (EU) 2024/1860 amended the IVDR in July 2024 and took effect on January 10, 2025. It introduced two critical changes:
-
Extended transition periods for legacy IVDs. If your device was certified under the old IVDD directive, you have additional time to transition to IVDR compliance. For Class B and sterile Class A devices, the deadline is 26 May 2027 if conditions are met.
- UDI and EUDAMED integration. Your QMS must now track unique device identifiers and ensure data flows to EUDAMED. Four modules (Actor Registration, Device Registration, Notified Bodies and Certificates, Market Surveillance) are live as of May 2026. Clinical Investigations and Vigilance modules arrive later in 2026.
What is proposed: Commission proposal COM (2025) 1023 (16 December 2025)
Published 16 December 2025, this proposal was under review (until 18 March 2026) and aims to simplify, not deregulate. The key elements are:
-
Proportionate documentation. This includes a narrower scope for Summary of Safety and Performance (SSP). It is required only for devices undergoing full technical documentation assessment. PSUR update frequencies extended for lower-risk devices.
- Risk-based certification. Certificates would be valid based on risk profile rather than fixed five-year expiry. This removes artificial recertification cycles.
- Streamlined audits. Periodic audits become risk-based rather than routine. Notified body surveillance focuses on high-risk areas.
- AI transparency. AI used in critical documentation must be documented with human oversight. This is now a regulatory expectation.
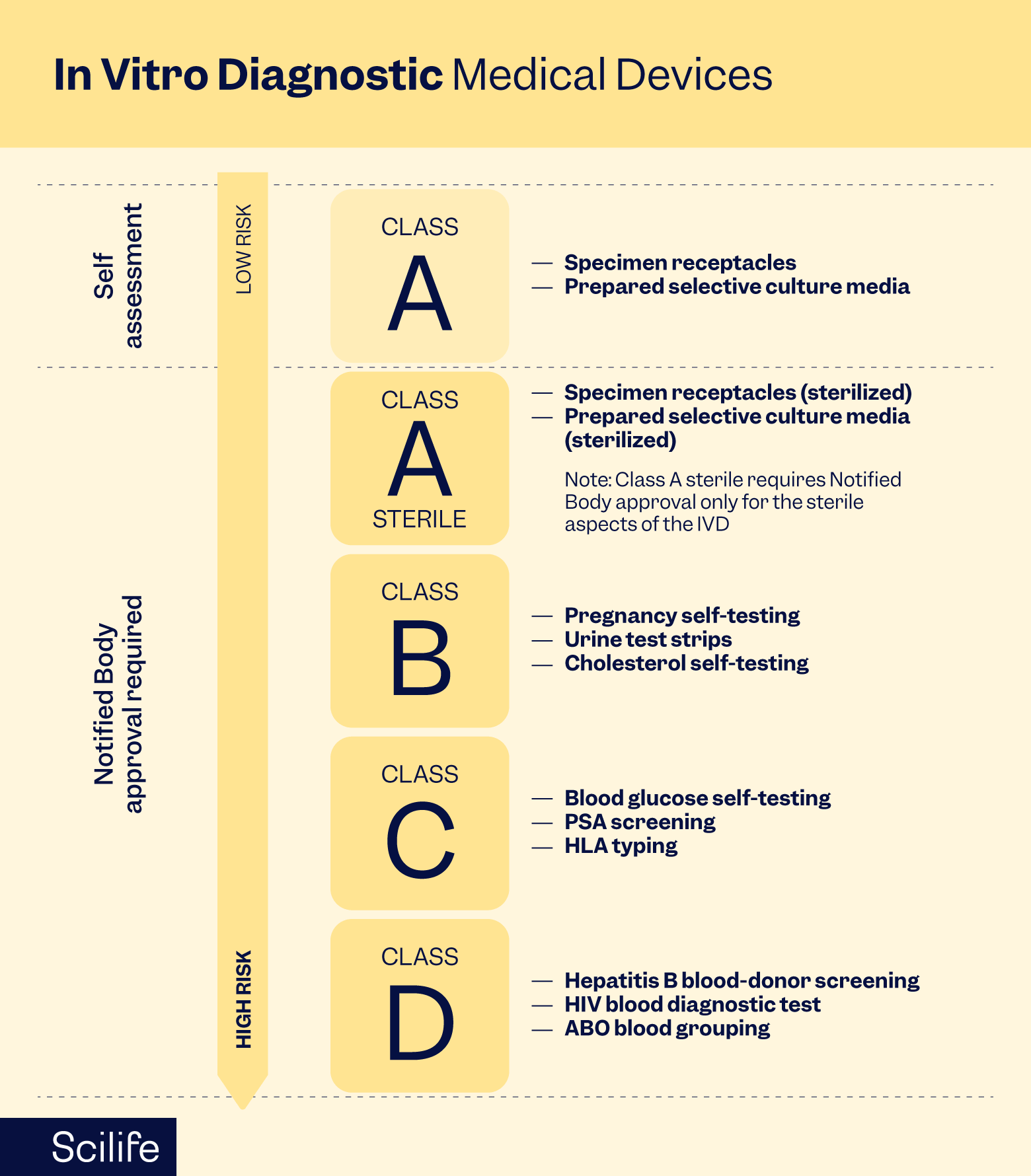
The 5 pillars of IVDR-compliant QMS
Establishing a QMS tailored to IVDR requirements is critical for compliance. It requires an adaptive framework tailored to your specific device risk classification. It involves defining organizational roles and responsibilities, documenting procedures, implementing risk management processes, and ensuring traceability throughout the device lifecycle.
At its core, IVDR QMS rests on five non-negotiable pillars. Each is essential. None can be skipped.
- Document Control: Maintain accurate procedures, work instructions, and technical documentation that support IVDR conformity. Ensure traceability across the device lifecycle. Include all data that supports your classification, clinical performance, and post-market surveillance strategy.
- Personnel and roles: Designate a Person Responsible for Regulatory Compliance (PRRC) who is permanently and continuously available. Define all roles and responsibilities. Provide training in QMS principles, risk management, and regulatory requirements. Competence must be verified and documented.
- Risk management: Identify potential hazards across the device lifecycle: biological, chemical, mechanical, and software-related. Assess severity and probability of harm. Implement controls that reduce risk to acceptable levels. Residual risk is documented and communicated to users. If AI tools assist in risk identification or assessment, document this and ensure human validation.
- Supplier and subcontractor management: Implement risk-proportionate oversight of suppliers, parts, and materials. When the safety profile of your finished product depends on third-party operations, the QMS must account for process controls at the supplier's production facility. Ensure that suppliers and subcontractors adhere to applicable regulatory requirements and provide necessary documentation to support the quality and safety of components and materials.
- Corrective and Preventive Actions (CAPA): Establish procedures for investigating non-conformities and complaints, such as implementing corrective actions to eliminate root causes, evaluating trends to catch emerging risks before they escalate, and also document continuous improvement activity.
Risk management in practice
Risk management is not a box to tick. It plays a pivotal role in ensuring the safety and performance of IVD devices throughout their lifecycle. Manufacturers must build comprehensive risk assessments to isolate potential hazards, evaluate the severity and probability of harm, and implement measures to mitigate risks to an acceptable level. It is an ongoing process that feeds into every QMS decision.
Key elements of risk management under the IVDR include:
- Hazard identification: Identify and characterize potential hazards specific to your device, including biological, chemical, mechanical, and software-related failure modes across the product life cycle.
- Risk assessment: Estimate the severity of harm that could result from each identified hazard and likelihood of occurrence.
- Risk control: Implement measures to eliminate or reduce identified risks to an acceptable level, such as design modifications, protective measures, or warnings and precautions that reduce risks. Test effectiveness.
- Risk communication: Clearly communicate residual risks to users, healthcare professionals, and regulatory authorities through labelling and instructions for use. Users must understand what risks remain.
- AI and risk documentation: If AI assists in hazard identification, risk assessment, or documentation, record the AI tools role, the human review process, and the final decision. Transparency is non-negotiable.
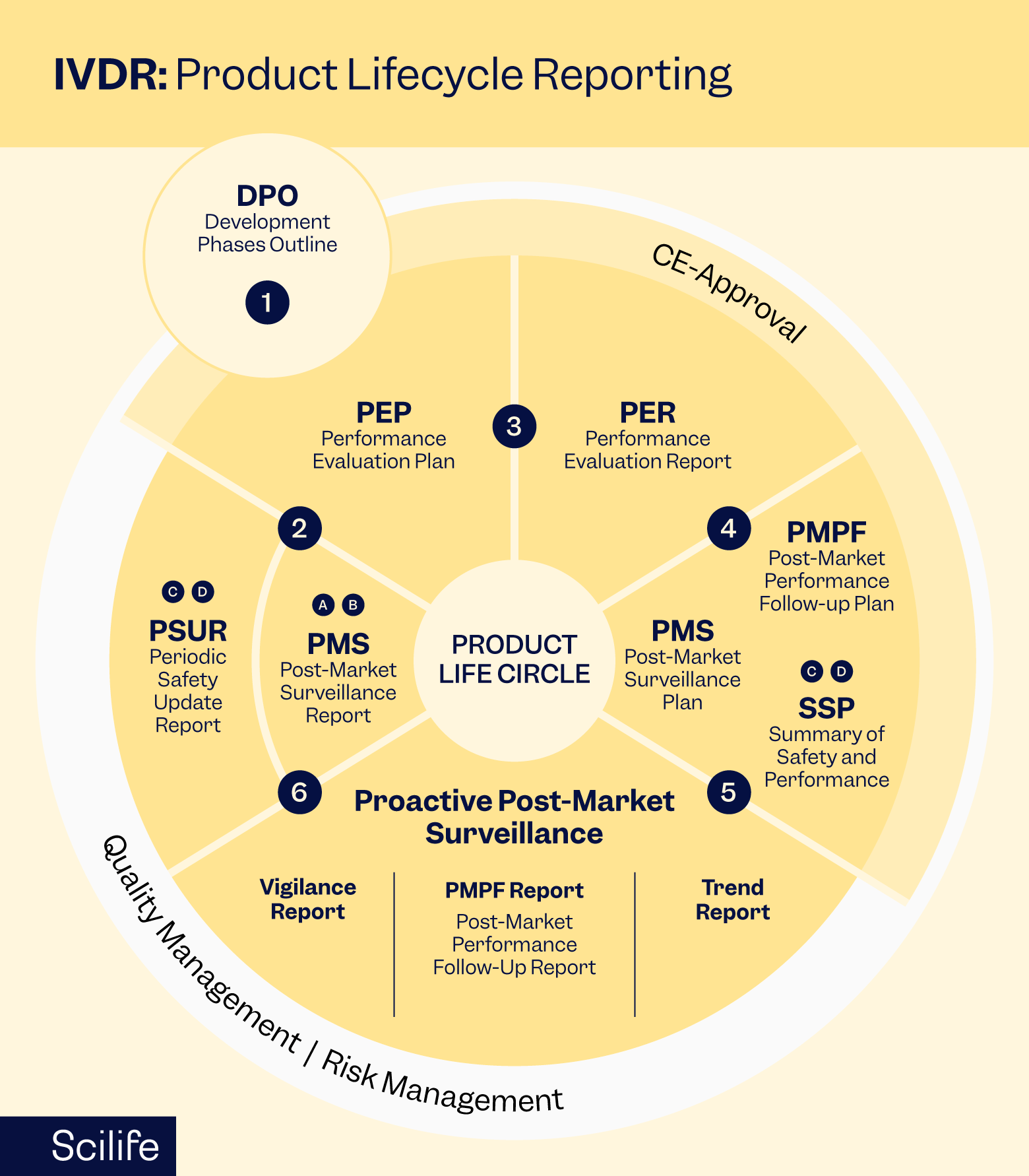
Post-Market surveillance in the EUDAMED era
Post-market surveillance (PMS) is essential for monitoring the performance and safety of IVD devices once they are placed on the market. It is no longer a separate archive. Instead, it is now integrated with EUDAMED, the EU device registry and vigilance system. Your QMS must reflect these integrations. Check our practical EUDAMED guide.
Key components of PMS include:
- Vigilance reporting: Promptly report any serious incidents, (deaths, serious deterioration in health, malfunctions) to competent authorities without undue delay. Use EUDAMED to submit reports. Delays harm patient safety and invite enforcement action.
- Post-market performance follow-up (PMPF): For Class C and D devices, conduct post-market performance studies to verify device claims. Document findings in your Performance Evaluation Report (PER). EUDAMED will streamline this process as modules come online.
- Periodic safety update reports (PSURs): Manufacturers must prepare and submit PSURs at regular intervals for Class C and Class D devices to summarize real-world safety and performance data, including any corrective actions taken. The December 2025 proposal would extend update frequencies for lower-risk devices, reducing cycles without lowering oversight.
- Trend analysis: Monitor statistically significant trends in complaints, non-serious adverse events, and malfunctions. These may signal emerging risks that require investigation and corrective action. These signal emerging risks. Act on trends proactively. Document all investigations.
- EUDAMED integration: Ensure your QMS captures data in formats EUDAMED accepts. Use UDI consistently across all registrations and reports. Prepare for upcoming vigilance module go-live. Synchronise internal systems with EUDAMED workflows.
7 practical ways to be compliant
Achieving full compliance with IVDR requirements requires a proactive and comprehensive approach. Here are some tips and recommendations to help you navigate the regulatory landscape effectively:
- Engage with notified bodies: Collaborate with notified bodies early in the process to gain insights into their expectations and requirements for IVDR certification.
- Transition to EUDAMED: Implement structured data capture now, and transition QMS files into EUDAMED-compatible formats. Stay informed of EUDAMED timeline updates. Monitor European Commission guidance regularly.
- AI use: Document AI use transparently. If AI generates, reviews, or validates critical documentation, record the tool, the human review process, and the final rationale.
- Utilize expert panels early: For Class C and Class D innovations, leverage the scientific advisory framework. Manufacturers can obtain early scientific advice from expert panels to confirm their clinical performance paths and performance evaluation methods before lodging high-stakes certification submissions.
- Suppliers management: Conduct supplier audits on risk profile. High-risk suppliers should get more frequent audits; low-risk suppliers less so.
- Invest in training: Ensure personnel understand why QMS processes are followed, not just what they are and how to complete them.
- Leverage technology: Explore software solutions and digital tools to streamline QMS processes, facilitate documentation management, and enhance traceability and transparency.
Conclusion
Compliance with the IVDR is a fundamental requirement for manufacturers of IVD devices seeking to launch or maintain products on the European market. By establishing a robust data-driven QMS, implementing effective risk management strategies, incorporating streamlined personnel requirements, and following best practices for compliance, manufacturers can fulfill compliance obligations confidently and ensure the safety and efficacy of their products.
Stay proactive. Engage with regulators and notified bodies early. Document your decisions transparently. Use EUDAMED modules as they become available. Treat AI as a tool that must be transparent and auditable, not as a replacement for human judgement. These practices keep you compliant today and ready for tomorrow.