In Vitro Diagnostics (IVD)
In Vitro Diagnostics (IVD) help to detect diseases and other health conditions.
What Are In Vitro Diagnostics?
IVD products are devices and systems used to diagnose, treat, or prevent health conditions. They are meant to be used in the collection and examination of biological samples like blood, saliva, or tissue. Samples may be taken from inside the nose or the back of the throat, or from a vein or fingerstick.
It’s important to note that IVD products are noninvasive. However, they are still considered medical devices and are therefore subject to the same controls. Medical devices consist of any instrument, appliance, software, or related device intended to be used for human beings for the purpose of:
- Diagnosing, preventing, monitoring, or treating disease or injury.
- Supporting or sustaining life.
- Regulating conception.
- Disinfecting medical devices.
- Providing information for medical purposes by collecting biological samples from humans.
Manufacturers of In Vitro Diagnostics must meet the regulatory requirements of the European Union, the United Kingdom, and the U.S. depending on the market(s) they wish to penetrate.
These requirements include the In Vitro Diagnostic Regulation (IVDR) (EU) 2017/746 in the EU and the UK Medical Devices Regulations (UK MDR) 2002 in the UK. In the United States, IVD products are defined in section 201(h) of the Federal Food, Drug, and Cosmetic Act (FD&C Act); these devices may also be subject to section 351 of the Public Health Service Act, as well as categorization under the Clinical Laboratory Improvement Amendments (CLIA) of 1988.
According to the IVDR, an IVD product is defined as “any medical device which is a reagent, reagent product, calibrator, control material, kit, instrument, apparatus, piece of equipment, software or system, whether used alone or in combination, intended by the manufacturer to be used in vitro for the examination of specimens, including blood and tissue donations, derived from the human body…”
Experts estimate that approximately 70% of all clinical decisions are made using IVD products.
Examples of IVD Products
Pregnancy tests, COVID-19 tests, and HIV tests are examples of IVD products. Other examples of IVD devices include:
- Cancer diagnostics
- Blood glucose monitoring systems
- Blood grouping devices
- Human genetic testing devices
- Immunoassays
- Hepatitis tests
- Self-tests and near-patient testing devices
- Coagulation test systems
- Urine test strips
Receptacles manufactured for medical specimens are IVD products as well. According to the World Health Organization, over 40,000 products are available for IVD testing today. These may range from traditional laboratory tests to point-of-care tests.
IVD Classification
In the United States, the FDA classifies all medical devices—including IVD products—as Class I, Class II, or Class III. The device’s classification will vary based on the risk involved and the level of regulatory control needed to guarantee the product’s safety. Accordingly, the IVD classification will determine the premarket process the manufacturer needs to follow in order to bring their product to market.
- Class I products are considered low to moderate risk and require general controls.
- Class II products are considered moderate to high risk and require general controls and Special Controls.
- Class III products are considered high risk and require general controls and Premarket Approval (PMA).
It’s important to note that as the device class increases, the regulatory controls increase as well. Class I IVR devices are subject to the least regulatory control, while Class III IVR devices feature the most stringent requirements. Life Sciences companies looking to bring their In Vitro Diagnostic products to market in the United States can refer to the FDA’s “Classify Your Medical Device” resource.
Looking to ensure regulatory compliance in the European Union? The IVDR was upgraded in 2021 to include improvements such as:
- Clear obligations for manufacturers, importers, and distributors involving device traceability, registration, and verification of proper labelling. This includes a traceability system founded on a unique device identifier (UDI).
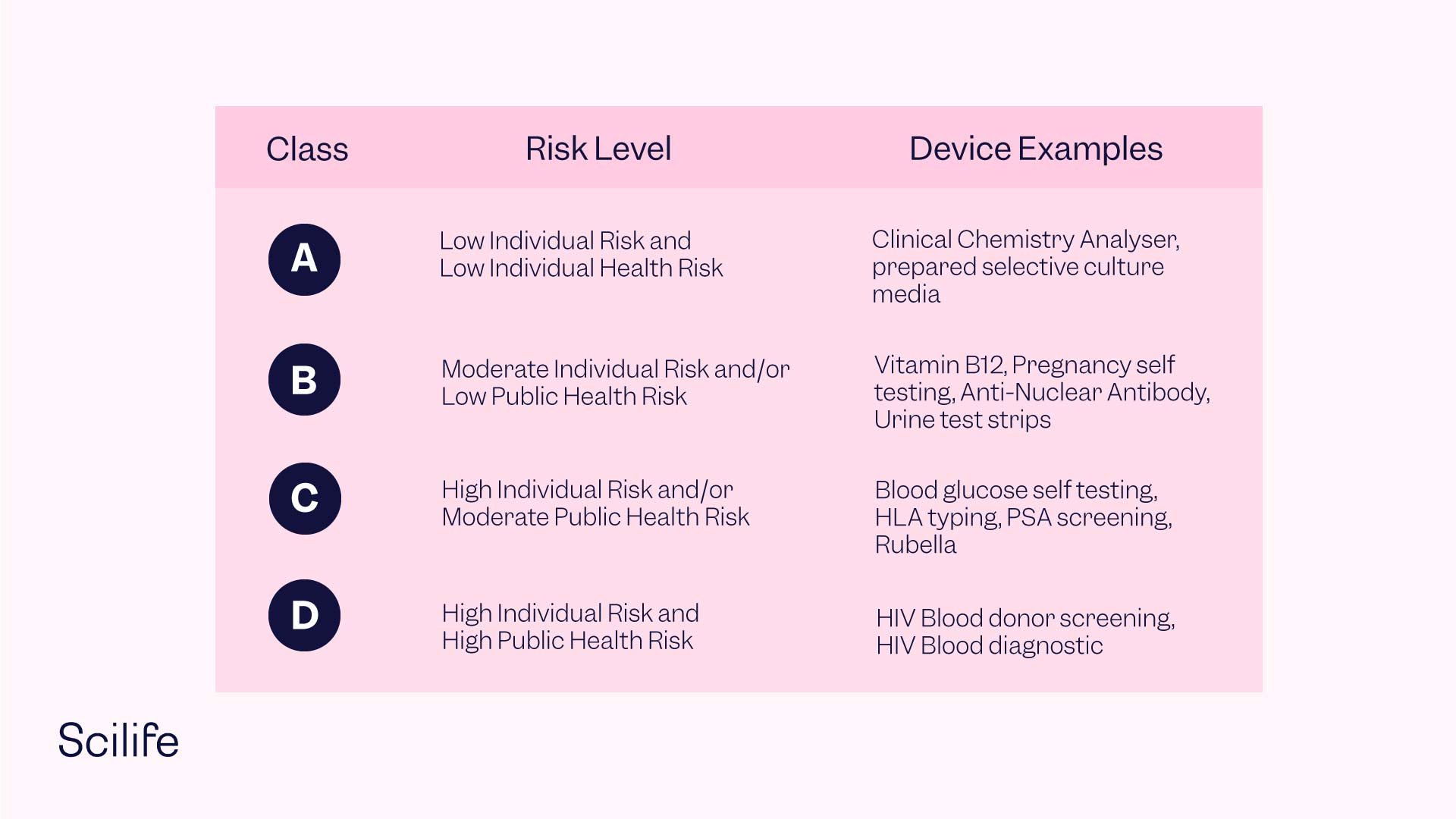
- Introduction of a classification composed of four risk classes. Class A devices feature low individual and public health risk, Class B devices feature moderate individual and/or low public health risk, Class C devices feature high individual risk and/or moderate public health risk, and Class D devices feature high individual risk and high public health risk.
- Stricter controls for high-risk IVD devices.
- Guaranteed oversight of notified bodies (or independent third-party conformity assessment bodies) by reinforcing a specific set of criteria.
- Greater transparency with a comprehensive database on medical devices in the EU (called EUDAMED).
- Reinforced clinical evidence and performance evaluation regulations, with more coordination among EU countries—especially where market surveillance is concerned.
- Stronger manufacturer post-market surveillance requirements.
- Specific requirements for “in-house devices,” or IVD products manufactured and used within the same institution.
Getting an IVD product to market safely and effectively is key—and understanding the regulations in place is an ideal first step.
Conclusion
IVD products are a critical line of defense against various health conditions. The safety and quality of In Vitro Diagnostics is essential, which is exactly why manufacturer regulatory compliance is so important.
Additional resources

How to Implement the Continuous Improvement Cycle | Scilife
Even an organization with stellar leadership and a solid core of employees experiences hiccups from time to time. Despite having assembled all the ...
How to assess and enhance your Quality Management Maturity | Scilife
As the life sciences industry becomes increasingly regulated and competitive, quality management has become more vital than ever. Are you confident ...

8 Best QMS Software for Life Sciences (2026 Comparison)
The right electronic Quality Management System (eQMS) can help strengthen your compliance processes and build a culture of quality within your ...

How to write a good quality plan for medical devices | Scilife
In life sciences, especially if you’re in the medical device industry it becomes harder to manage projects in accordance with your company’s quality ...
Turn quality into your brightest asset with Scilife

