

ALCOA was born on a crowded slide in the early 1990s. An FDA officer called Stan Woollen ran out of room while preparing a presentation, so he typed five letters as a memory prompt: Attributable, Legible, Contemporaneous, Original, Accurate. The acronym stuck.
Three decades later ALCOA sits at the heart of how inspectors judge your records.
Data integrity is now one of the most frequently cited observations in FDA GMP warning letters. If you lead quality, validation, CSV, or IT in a GxP environment, a loose grip on your data is the fastest route to a finding.
In this post we’ll explain what a data integrity risk assessment is, how it differs from a CSV impact assessment, when to run one, and a simple, repeatable method you can use, with ALCOA+ examples included.
Key takeaways
What is a Data Integrity Risk Assessment (DIRA)?
A data integrity risk assessment is a documented, risk-based exercise. You map the processes that produce or capture your data, identify each format and its controls, and record the data's criticality and inherent risk.
The MHRA GxP data integrity guidance names it directly as DIRA and presents it as an example of a suitable approach for keeping a system in an acceptable state of control, supported by a documented rationale.
The purpose of DIRA is focus: having confidence in the quality and integrity of the data behind your decisions on patient safety and product quality, and being able to reconstruct what happened when you are challenged.
You cannot guard every field with equal force, and regulators do not expect you to. The effort should match the impact of a failure on the patient, the product, or the environment. A DIRA tells you where to spend that effort, and where you can ease off.
DIRA vs. CSV impact assessment: what’s the difference?
Among the most confusing differences I see in the life sciences is between DIRA and CSV impact assessments.
-
A CSV impact assessment is system-centric. It askƒs one question:
-
Is this system GxP-relevant, and how much validation does it need?
-
It scopes the validation effort and the change.
-
-
A DIRA is data-centric. It asks a different question:
-
Across the life of this data, where could it be lost or quietly changed, and would we notice?
-
DIRA and CSV impact assessment overlap, but they are not interchangeable. A fully validated system can still produce vulnerable data, because automation lowers risk without removing it.
I once reviewed a chromatography setup that had sailed through validation, yet shared logins meant nobody could say who had reprocessed an integration. The system was valid, but the data would not pass an audit.
What “data integrity” really means: applying ALCOA+ in practice
Data integrity is the degree to which data stays complete, consistent, accurate, trustworthy, and reliable across the whole data life cycle.
The MHRA GxP data integrity guidance sets out ALCOA+ as the benchmark, turning that idea into nine practical attributes. PIC/S and the WHO apply the same ALCOA+ attributes, so the expectation travels with you across regulators.
Here is what each principle looks like on the floor:
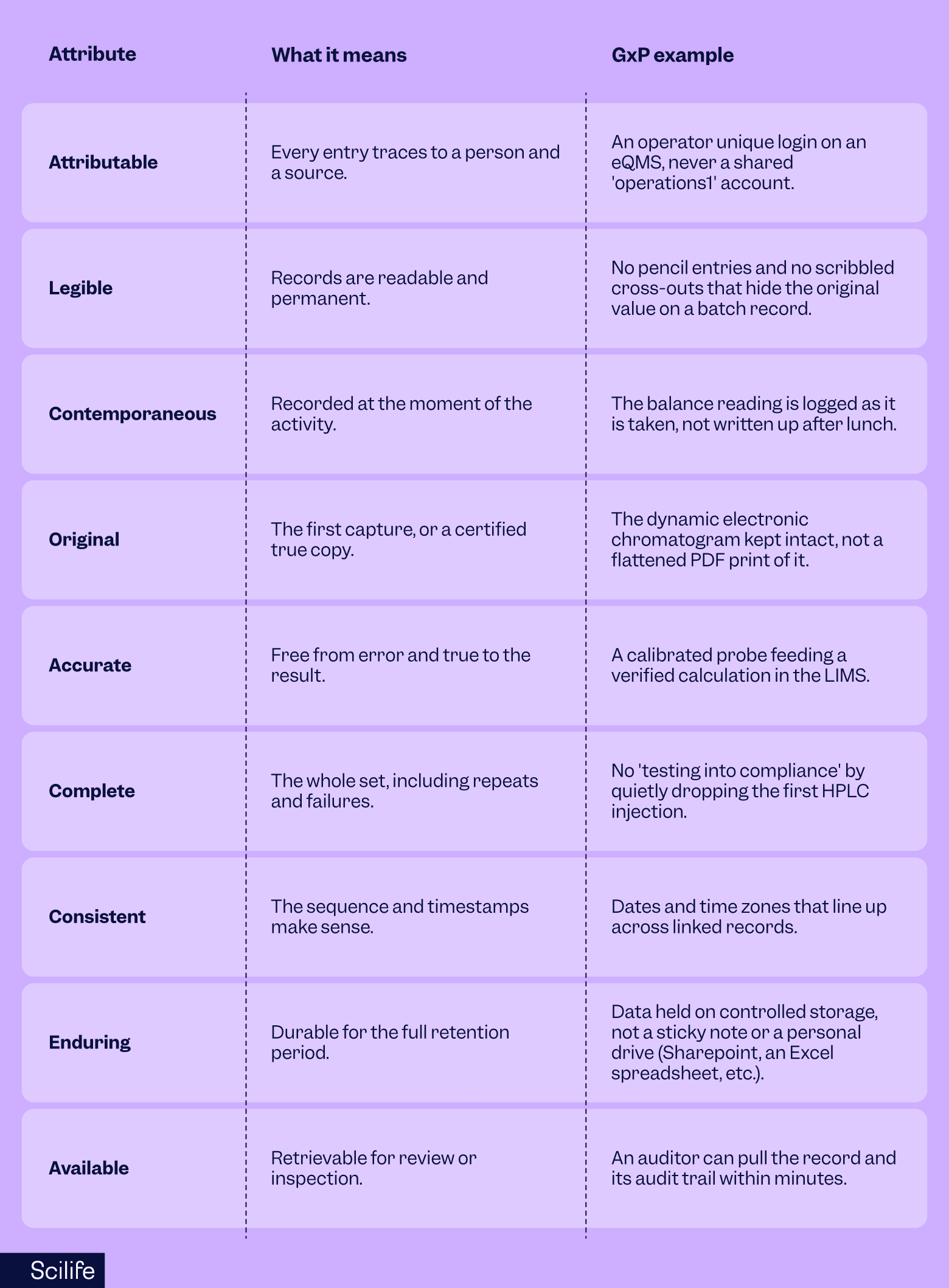
ALCOA+ extends the original five with Complete, Consistent, Enduring, and Available. The MHRA is clear that the expectation is identical whichever acronym you use. The plus simply spells out what good data governance already implies.
When should you perform a data integrity risk assessment?
The first thing you should do is to ensure that your data integrity risk assessment is a living one and not a one-time project.
Every time the risk picture changes, a risk assessment should be conducted or refreshed:
-
A new system or process goes live, or you onboard a new supplier or cloud service.
-
A major change lands: an upgrade, a configuration change, or a new interface between systems.
-
A periodic review falls due, so the assessment keeps pace with how the system is actually used.
-
An audit or inspection finding points at how you handle data.
-
Migrating data presents many challenges, especially maintaining the records' full meaning.
-
You move between paper and electronic, in either direction, which never removes the need for controls on its own.
The DIRA method: a simple, repeatable approach
Whenever I need to solve a problem, I turn to the DIRA method. It scales from a single instrument to an enterprise platform, because the logic is the same at any size.
-
Describe the purpose of the system or process. Describe the data and records that carry GxP weight. Vendor validation alone isn't enough; you must understand how the system fits into your own workflow.
-
Map the data flow. Follow each record through its life cycle: create, modify, review, approve, report, archive. This step, often called GxP mapping or GxP data mapping, is the backbone of the exercise, and it forces you to understand your file structure.
-
Classify data criticality. Rank records by how much they influence quality or safety decisions.
-
Identify the inherent risks. At each step, ask how data could be deleted, amended, or excluded without authorisation, and how likely you are to detect it.
-
Assess existing controls and residual risk. Weigh technical controls such as access rights and audit trails against procedural ones, and be honest about what is left over.
-
Prioritise, document, and act. Rank remediation, accept residual risk where justified, communicate it to management, and review it. Where long-term fixes take time, implement short-term mitigations in the meantime.
The map is only ever as good as the people who know where the weak points really sit. So, bring subject matter experts into the room. Regulators expect it. The MHRA names the inclusion of subject matter experts as an enabler of data integrity.

Recommended learning:
Data integrity in pharma: FDA, EU GMP and cGMP compliance explained.

What auditors expect from a data integrity risk assessment
Auditors and inspectors are not looking for a glossy form. They need evidence that you understand your data and have made informed decisions. So the first thing they will probe is whether your system is documented and whether you can explain the rationale behind the controls you chose.
A clear justification of why you did something carries far more weight than a polished template with no critical thinking behind it.
From there, expect them to follow the data. They will want your data flow maps, because those show you actually where records live and where they move.
They will look for evidence of critical data and inherent risk rather than vague assurances, and they will expect audit trail reviews to be risk-based, rather than applied blindly to every click.
Then they turn to what you did about it. Note that a credible DIRA ends in action:
-
Remediation plan with prioritised steps
-
Residual risk that has been accepted and signed off by management
-
Short-term mitigations running while the longer fixes are delivered
A finished assessment that nobody has revisited in three years is its own red flag.
Common mistakes in DIRA (and how to avoid them)
Most failed DIRA assessments fail because of a handful of predictable traps, the kind that look reasonable while you are in them and obvious once an inspector points them out.
Here are the ones I see most often, and how to stay clear of them.
-
Viewing DIRA as a one-time event. The risk picture is not static. A new interface, a software upgrade, a change in how the team actually uses the system, any of these can move the goalposts. If your assessment was signed off two upgrades ago, it is describing a process that no longer exists. Tie it to change control so it refreshes when the system does.
-
Confusing DIRA with a CSV impact assessment. Validating the system is not the same as protecting the data. A CSV impact assessment tells you how much validation a system needs; it does not tell you whether someone can overwrite a result downstream. Both should be run, and you should be clear in your head which one answers which question.
-
Assessing only the software. People, training, and procedures carry as much risk as the software itself. Automation lowers risk but never removes it, and the moment a human can influence what gets recorded, reported, or retained, your weak point is usually the operator and the SOP, not the code. So, here’s a key takeaway: Map the whole process, not just the screen.
-
Mapping only the ideal flow. It is easy to map a process when you assume everything goes right. But the real risk lies in the exceptions, and in an eQMS those exceptions are everywhere. Think of the deviation that gets reopened and quietly re-dated, the CAPA due date pushed back with no record of who moved it, or the document that skips a review step because someone holds rights they should not have. The same goes for a training record marked complete without the assessment behind it, or an audit trail that nobody reviews because the report was never configured to surface changes. These are the routes auditors look for, so map them on purpose. Trace what happens when a record is corrected, escalated, overridden, or rolled back, not just when it sails through the workflow for the first time.
-
No owner for residual risk. If nobody signs for it, nobody manages it. A residual risk that sits in a spreadsheet with no name against it has not been accepted, it has been ignored, and that is exactly the gap an auditor will find. In an eQMS this is easy to close. Give every open risk a named owner, a clear decision, and a review date, then link it to the records that keep it visible: a CAPA, a change control, or a periodic review task that reminds someone before the deadline passes. The point is not just to document the risk, but to make sure a real person has to look at it again and answer for it in front of management.
-
Trusting the validation certificate. I have watched teams treat a validated state as a comfort blanket, right up to the moment an auditor asked who could switch off the audit trail. Validation tells you the system worked as intended on the day it was tested. It says nothing about who holds admin rights today, whether shared logins have crept back in, or whether someone disabled a control during the last upgrade. In an eQMS these things drift quietly: a new starter is granted more access than the role needs, a configuration change goes live without a fresh assessment, an audit trail setting gets switched during a patch. Remember, validation is the starting line, not the finish. Keep checking who can do what, and confirm the controls you validated are still switched on.
Using templates and tools to streamline DIRA
Consistency is what separates a credible DIRA from a pile of one-off spreadsheets. I recommend preparing a data integrity risk assessment template, which gives every team the same columns to work through:
-
Data and format
-
Process step
-
Inherent risk
-
Probability of occurrence
-
Severity of harm
-
Detectability
-
Risk scoring (quantitative or qualitative RPN)
-
Risk control: risk acceptance or risk reduction
-
Risk communication
-
Mitigating action (owner, and action)
A shared template standardises the thinking. Using one template will allow assessments to be compared across systems and teams, and a reviewer will know just where to begin.
By the time the template is developed, an eQMS can scale it. This means the template resides within the eQMS rather than in an external drive.
Electronic templates are really useful because each risk can be linked to the source records. A change control can reopen the assessment whenever the system changes. A CAPA can track each remediation through to closure. An audit trail review can run as a recurring scheduled task, and a periodic review can prompt someone to revisit the whole thing before it goes stale. Nothing sits forgotten in a shared drive, because the system keeps putting it back in front of a real person. For the wider context this exercise sits inside, see our ultimate guide to computer system validation in pharma.
Conclusion
Strip away the acronyms. Data integrity risk assessment is a simple promise. You know where your data could fail, and you can decide what to do about it. That is how you protect the records, your decisions. Your patients depend on it, and it is how you walk into any inspection without panicking.
Start small. Map one critical process, run it through ALCOA+, and you will quickly see the gaps you have been living with. Then move on to the next one.
The goal is to turn DIRA into a living, auditable process rather than a document that ages in a drawer.
If you want to see how a digital eQMS keeps validation and data integrity connected and under control, explore the tools at Scilife, or go deeper in the Scilife Academy.





