
Medical devices have one purpose: to improve the life of patients. They are developed with this purpose in mind, and the entirety of regulatory compliance is designed to ensure that medical devices remain safe and perform as intended throughout their lifetimes.
But how can medical device manufacturers be sure they are actually improving lives? Through usability and labeling.
Usability
Usability for medical devices is a part of risk management and refers to how people use medical devices. Usability looks at the interactions between the indicated users of a device in the intended environment and is defined as human factors engineering (HFE).
The FDA defines human factors engineering as
"The application of knowledge about human behavior, abilities, limitations, and other characteristics of medical device users to the design of medical devices, including mechanical and software-driven user interfaces, systems, tasks, user documentation, and user training to enhance and demonstrate safe and effective use."
HFE considerations in the development of medical devices involve the three major components of the device-user system: (1) device users, (2) device use environments and (3) device user interfaces. The interactions among the three components and the possible results are depicted graphically in Figure 1.
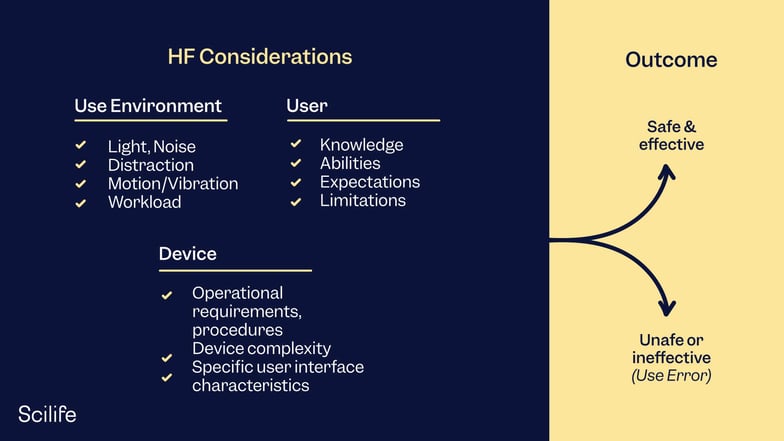
Figure 1: Interactions among HFE/UE considerations result in either safe and effective use or unsafe or ineffective use.
All medical devices must have a certain degree of usability and ease of use. Some devices are used exclusively by health professionals, while others are used both in clinical settings and at home by potentially untrained patients or caregivers. For this reason, designing a device that considers both populations is critical. While it might be clear to engineers and developers how a medical device should be used, healthcare professionals or patients might not have the same knowledge and considerations, which can lead to user error or even harm caused to the patient. Usability testing should be performed on anyone who is part of the use of a medical device, from patients to clinicians to sterilizers and service engineers.
Usability testing seeks to identify potential causes of user error and harm and mitigate them. In particular, complex devices, drug-device combination products, and life-sustaining products need thorough usability testing to ensure the device's user interface and design features won't cause adverse events. Use errors are relatively common in devices such as infusion pumps, automatic electronic defibrillators, ventilators, and drug auto-injectors, causing a variety of uses, from overdoses to improper deliveries to delayed treatment. Usability testing can help mitigate these issues when performed correctly.
Usability Testing and Procedures
Usability research examines how human operators interact with medical devices. It's essential to test everyone involved, including patients, clinicians, and those responsible for device maintenance and sterilization. Usability testing is generally divided into formative and summative assessments.
Formative assessments are used early in the design and development process to point out any design problems, create a functional user interface, and identify whether the functionality of a device is simple or complex.
Summative assessments are employed late in the process to ensure that any issues or problems related to the user experience have been identified and resolved and that health professionals and users won't commit errors that can cause harm during the use of the device. Usability testing has different requirements in different markets: for example, summative testing requires 15 participants in the FDA and 12 under the EU MDR. Likewise, many manufacturers use IEC 62366-1 Application Of Usability Engineering To Medical Devices, but different markets might require testing to other usability standards.
The EU Medical Device Regulation (2017/745, MDR) requires a complete analysis of usability risks and mitigation as far as possible - all the measures for identified risks related to usability must be defined, and risks associated with reasonably foreseeable misuse must be evaluated. Specific risks for various categories, such as inadequate ergonomic features, ergonomics of displays, and instructions for use, must be met for any device that falls under the categories in Annex I General Safety and Performance Requirements.
The FDA also expects specific methods for analyzing usability risks and planning and documenting usability testing to ensure the safety and efficacy for intended users, uses, and use environments.
Usability definitions and requirements are also mentioned in ISO 1384:2016 Medical devices — Quality management systems — Requirements for regulatory purposes and ISO 14971:2019 Application of risk management to medical devices.
Importance of Usability Testing
Usability testing is a critical step in the lifecycle of medical devices. Neglecting it can lead to issues requiring costly redesigns and delays in FDA/CE submissions. On the other hand, excessive testing can strain budgets. Striking the right balance is crucial. Usability testing not only aids FDA/CE approval but also reveals flaws and challenges. Addressing minor issues early on prevents them from becoming major concerns during device design and development.
Regulatory Frameworks For Usability Testing
Usability testing for medical devices is guided by specific regulatory frameworks, tailored to different markets. In the US, the FDA provides detailed guidance on human factors and usability engineering, outlining testing requirements and referencing the recommended adherence to IEC 62366 standards. Internationally, IEC 62366-1:2015 is a significant standard for usability engineering in medical devices. It addresses risk assessment, mitigation, and even identifies potential dangers from unusual device usage.
Europe's medical device regulation (EU MDR) incorporates usability requirements within its General Safety and Performance Requirements. Usability activities and validation must be documented in a manufacturer's technical file, with emphasis on gathering and analyzing usability data for post-market monitoring and evaluation. Regulatory organizations, including the FDA and European Commission, adhere to the same IEC 62366-1 standard for usability.
Conducting and Reporting Usability Testing
Specialized skills are crucial for conducting successful usability tests. If using internal resources, training and staff proficiency are essential. Alternatively, outsourcing to an experienced partner in medical device human factors and validation testing is a reliable option.
It’s important to form a team comprising engineering, regulatory, training, and marketing managers from your company, collaborating with the testing company. This group effort will enable the effective identification of user categories and the development of participant screeners. FDA guidelines suggest testing at least 15 individuals for each user group, followed by a comprehensive report.
After testing, prepare a usability engineering report in a specific format for submission to the appropriate regulatory body. The FDA outlines eight sections the report should include, emphasizing the need to demonstrate safety and effectiveness for intended users, uses, and environments.
Usability testing examines factors like time, resources, and efficiency, while incident evaluation identifies potential causes and their roots. Testing should involve a minimum of 5 users, assessing the device's interface, design, and instructions for proper usage.
Software testing is equally important, as malfunctioning can occur. Rigorous vetting is required for medical device software to prevent run-time errors. Additional software applications can be used for debugging and enhancing medical device programs. By following these steps, you can ensure the usability and safety of your medical device.
Establishing Usability Engineering Procedures in QMS
Design control requirements are outlined in 21 CFR Part 820.30, an essential subsystem of your Quality Management System (QMS). The FDA quality system regulation implies the need for human factors and usability testing in several sections, such as design input, verification, and validation.
Design input should consider the needs of users and patients, while design verification ensures performance criteria are met. Design validation involves testing production units under actual or simulated use conditions, including software validation and risk analysis.
Usability engineering (UE) should be integrated into the design review process within your QMS. It is crucial to avoid treating design reviews as a last-minute activity, as this can make it difficult to address design mistakes later in the development process.
To ensure a thorough evaluation, it is recommended to conduct design reviews at the end of each development phase, at a minimum, and include human factors and UE considerations. By incorporating UE into design control, you can enhance the usability and quality of your medical device.
Labeling
Labeling for medical devices does not refer only to the label itself – any essential information about a medical device and its use provided by the manufacturer is included when we refer to medical device labeling. This includes instructions for use, brochures, manuals, marketing materials, etc.
Appropriate labeling prevents user error. It also ensures the user can use and benefit from the device safely. Typically, device labels on the device itself include manufacturer information, device information, unique device identifiers, expiration dates, and symbols and legends pertaining to the appropriate identification and tracking of a medical device. Most markets have similar label requirements, typically based on FDA or EU MDR label regulations.

On the other hand, instructions for use include the intended use of the device, precautions and warnings, contra-indications, special considerations, and intended application.
According to the FDA, labeling is defined as
"…all labels and other written, printed, or graphic matter; (1) upon any article or any of its containers or wrappers, or (2) accompanying such article' at any time while a device is held for sale after shipment or delivery for shipment in interstate commerce." In this instance, accompanying means any physical documentation associated with the device, such as posters, tags, pamphlets, booklets, directions, instruction sheets, etc.
Labeling for medical devices is essential to convey the intended use and potential risks associated with the product design. The FDA's Guidance on Medical Device Labeling recommends various approaches to enhance the effectiveness of labeling:
- Integrating warnings and precautions within the context of tasks and hazards.
- Using concrete language instead of abstract terms and technical jargon.
- Keeping the content concise and using simple language.
- Using straightforward language that clearly communicates risks, such as "Contact with this product will produce severe burns" instead of "Contact with this product will result in serious injury."
Usability testing is another way to improve the usability of product labeling. It is a part of product verification and provides valuable data on user interactions, error rates, and any difficulties encountered while using the device. By incorporating usability testing, the labeling can be optimized to effectively communicate important information to users.
The EU MDR does not define labeling as clearly as the FDA but does require instructions for use to be provided with each device. Furthermore, the following article: Labeling Requirements for Medical Devices describes the requirements for labeling, instructions for use, and risk information, all of which are outputs of usability engineering.
How Usability and Labeling Overlap
It is clear to see how usability and labeling work together to ensure the best possible user experience. Usability looks to reduce user error and harm through design and user interface testing. At the same time, the device labeling clearly defines the intended use of the device and any warnings and precautions needed to use the device safely. Essentially, the device labeling communicates to the user how they are supposed to use the device safely, what they should avoid doing, and which situations can cause harm. It provides clear instructions to the user, ensuring they are not harmed due to incorrect use.
While it might be obvious how to use simpler devices such as splints or monitors, particularly with higher-risk devices, many of the instructions in the manuals are derived from usability testing and validation. Many instructions for use could be more effective and easily understandable by laypersons. Similarly, more health professionals need to read the instructions before using a device. Likewise, usability testing can assess the efficacy of your labeling before launch and enhance its value to your users.
Conclusion
The relationship between usability testing and labeling in medical devices is crucial for ensuring patient safety and improving the overall user experience. Usability testing helps identify potential user errors and design flaws, allowing manufacturers to mitigate risks and enhance the device's usability. By involving various stakeholders, including patients, clinicians, and maintenance personnel, usability testing can address the needs of different user groups and environments.
Proper labeling, including instructions for use, warnings, and precautions, plays a vital role in conveying essential information to users and minimizing the potential for misuse or harm. Following the guidelines provided by regulatory bodies such as the FDA and EU MDR, manufacturers can optimize labeling by integrating warnings, using clear and concise language, and aligning it with the device's intended use.
The overlap between usability testing and labeling is evident in their shared goal of promoting safe and effective device usage. Usability testing helps inform the development of clear and user-friendly instructions, while labeling ensures that users understand how to use the device safely and avoid potential risks. By integrating usability engineering into the quality management system and considering usability throughout the design and development process, manufacturers can enhance the usability and quality of their medical devices, ultimately improving patient outcomes. At Scilife, improving patients’ lives is a vital part of our mission.
If you work in the medical device sector, have a look at how Scilife Smart QMS can support you!





