

Developing and bringing a medical device to market is a scary process. Not only do you have to come up with a great idea or design improvement and develop a working prototype, but you must also ensure the device is safe and performs as intended in any indicated situation.
Potentially the most daunting task of medical device development is the regulatory aspect. Ensuring regulatory compliance can delay any medical device launch if not done adequately and in good time.
Here, we review what it takes to bring a medical device to market once inspiration has struck and you’ve come up with a great idea, from formulating a business plan to post-market responsibilities.
And if you're looking to get ahead of the regulatory curve and avoid surprises down the line, our QARA guide to medical device quality management systems breaks down the essentials of building a compliant, inspection-ready QMS.
Business Plan
Ideas for potential medical devices can come from anywhere – medical device research and development departments, clinicians wanting a better solution for their patients, or engineers trying to fix problems on existing devices. Many medicines and devices have sprung from a current device being used differently than intended.
The obvious next step for someone with a good idea is to develop the device design and start prototyping right away. This is especially true for health professionals eager to make life easier for their patients.
The medical device industry is a heavy beast to dance with, though, and in the case of developing medical devices with the eventual plan to bring them to market, there are a few recommended steps to go through first.
Market Research
Maybe the most essential step of medical device development, market research, is often under-prioritized. Even the best idea will not go anywhere unless there is a market demand for it.
The easiest way of determining if your device might be successful is to investigate any existing competitors. Maybe your device is one-of-a-kind, and no competitors exist, in which case it is helpful to investigate why. Did no one ever think of your device idea? Or is it not viable in the market? Some devices are so niche that acquiring them is not worth it when other devices with similar technologies can be used instead. Some cultures frown upon certain devices or solutions and hesitate to use them. Some markets are simply saturated with alternative technologies.
If competitors are already on the market, you can look into their device design, revenue, and sales to get an idea of the size of the specific device market. It is also worth asking yourself how your device differs from others on the market, i.e., why should customers buy your product instead of the existing ones?
It would help if you also looked into the size of your potential target markets. Smaller or national markets might be easy to overview, but larger international markets will frequently require some research. The size of your target markets will determine potential revenue, regulatory priorities, and how much funding you can realistically raise.
A good idea is to summarize everything in a market research report. Market research reports can provide summarized overviews of existing technologies, competitor devices, and economic considerations. Coupling the market research report with a state-of-the-art report offers a complete image of the current market, as the state-of-the-art report can determine the most common materials used, different device indications, reviews on the clinical condition and its history, and alternative therapies based on scientific literature searches.
Project Timeline and Costs
With market research in the bag, you can begin working on your project timeline and the costs associated with every step of the development process.
Taking the time to develop a realistic budget is key. Every developmental stage requires different costs and solutions. Even though there will always be surprises or unexpected expenses, taking these into account from the beginning can avoid delays in the developmental timeline. Some funding channels depend on reaching certain milestones, so realistically mapping out the costs of each phase will also help you ensure continuous capital availability. The project budget should include various costs, such as regulatory compliance, materials, personnel and training, external consulting, product development and testing, manufacturing, clinical trials, any travel required, launch and marketing costs, and more. Many medical device manufacturers have had to postpone their product launch because of unexpected expenses or hidden surprises during testing and manufacturing, so adding a cushion for any unexpected costs can also be helpful.
The same goes for project timelines. Product development can take years, and in particular, regulatory compliance takes a very long time to get right. Even though everyone is eager to launch and make money on this great new device, taking the time to make realistic timeframes for each stage can end up saving everyone a lot of headaches.
Prototyping
Once you know there is a place on the market for your device, and you have the timelines and costs down, you can start developing and improving your prototype. The easiest way to create an initial prototype is 3D printing, which can help you produce a cheap prototype that can be easily modified. When you’ve settled on the finished design, you can have a more robust prototype built, either in-company or with an external prototype.
Your prototype will be the basis for feasibility studies to determine whether you should proceed with your device development project.
Regulatory Planning
You have a working prototype, and at this point, you’re probably ready to put everything you’ve got into developing your device and moving forward in the project. The last step in the business plan, however, might be the most complicated.
Regulatory planning can intimidate even the most experienced developer, and for good reason – it might be the most challenging part of putting a medical device on the market. It is also the aspect of device development most likely to delay your market authorization, so it is crucial to have an initial plan.
Here we will mainly discuss regulatory compliance in the context of the European Union (EU) and the United States of America (US). The principal applicable regulations are the European Medical Device Regulation (EU MDR, 2017/745), the European In Vitro Diagnostics Regulation (EU IVDR, 2017/746), and the Food and Drug Administration’s (FDA) Medical Device Regulation 21 CFR Part 800.
Defining the specific intended use of your device might not be considered the natural starting point for some manufacturers. Still, the intended use will determine your device classification, which in turn can determine the level of documentation required in each market. The intended use should define a device’s medical purpose, intended patient population, medical conditions, indications and contraindications, therapeutic area, and any warnings and precautions needed. The intended use will also help you establish user needs, design inputs, and other aspects of design and development. Something to look out for when drafting your intended use is to make sure the intended use is accurate and precise. Marketing departments tend to want to enhance the intended use for marketing purposes, but it can complicate classification in certain countries. Be especially careful with the term “diagnostic,” as it tends to have a massive impact on classification. If your device helps the health professional diagnose, but the intended use claims it diagnoses directly, some regulatory authorities will insist on a higher classification than you expected.
Classification is the next part of your regulatory planning. Classification is based on the risk profile of your device, i.e., level of invasiveness, potential harm, duration of contact, and intended use. Lower-risk devices typically won’t harm the patient if they malfunction or will cause very little harm. In contrast, higher-risk devices can cause serious impairment or even death in cases of malfunction or adverse events. Classifications can change from market to market but are generally similar, if not the exact same.
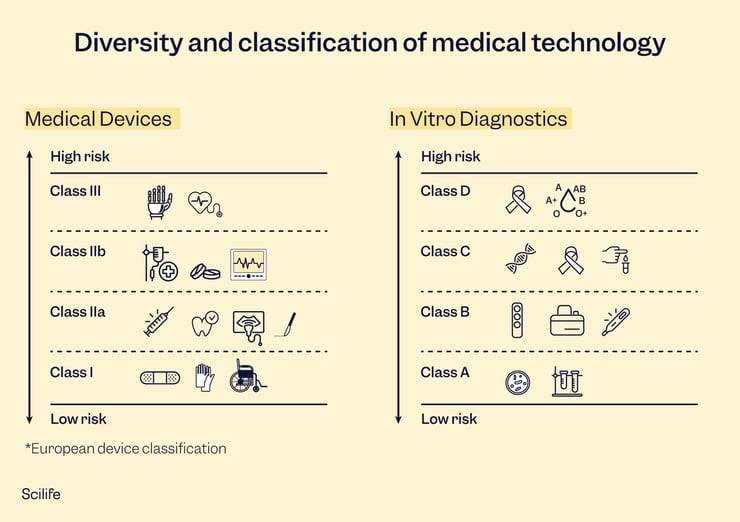
Choosing your target markets is another important step on the road to device development. You don’t need to launch in all your target markets at once, but it is good to have an idea of where you want to sell your device long-term. Larger markets, such as the EU and the US, work well as stepping stones to enter smaller markets worldwide, as some smaller markets will use an EU or US approval as leverage for faster approval. Your device classification will determine the path to approval in each market.
The last part of your regulatory planning is to consider your Quality Management System (QMS). A QMS is required to produce medical devices in almost all markets around the world and is a natural part of medical device manufacturing. Larger companies will already have a QMS in place, but smaller companies might need to set up a QMS from scratch. The best option for any medical device manufacturer is to comply with ISO 13485 Medical devices — Quality management systems — Requirements for regulatory purposes. While the US has their own QMS regulation, the 21 CFR Part 820, essentially all markets outside the US apply ISO 13485. The two regulations are quite similar and are constantly being aligned over time, so being compliant to either can significantly help you comply with the other.
Medical device marketing and industry trends
Bringing a medical device to market doesn’t end with regulatory approval—it begins a new challenge: achieving market adoption in an increasingly competitive and regulated landscape. While strong market research lays the foundation, a well-defined marketing strategy ensures your device reaches the right stakeholders and delivers real-world impact.
The shift toward data-driven and digital marketing
Medical device marketing has evolved significantly in recent years. Traditional sales-heavy approaches are being replaced by data-driven and digital-first strategies, where companies rely on analytics, real-world evidence, and targeted communication to engage healthcare professionals and decision-makers.
From educational webinars to omnichannel campaigns, medtech companies are now expected to deliver relevant, compliant, and personalized content throughout the buyer journey
Value-based messaging in a regulated environment
Unlike other industries, medical device marketing must strike a careful balance between innovation and compliance. Claims must be fully aligned with regulatory approvals, and messaging should clearly communicate:
- Clinical benefits and safety
- Economic value for healthcare systems
- Differentiation from existing solutions
As highlighted earlier, even small changes in intended use or positioning can impact regulatory classification, making alignment between marketing and regulatory teams essential.
Key industry trends shaping commercialization
Several trends are currently reshaping how medical devices are brought to market:
- Increased regulatory scrutiny: Global regulations such as EU MDR and FDA requirements are raising the bar for evidence and documentation.
- Rise of software and connected devices (SaMD): Digital health solutions are changing both product development and marketing narratives.
- Focus on post-market data: Real-world performance and post-market surveillance are becoming key marketing assets.
- Longer time-to-market: Companies must plan earlier and invest more in both compliance and commercialization strategies.
These trends reinforce the need for integrated workflows, where quality, regulatory, and commercial teams collaborate from the early stages of development.
Building a strong go-to-market strategy
To successfully commercialize a medical device, companies should:
- Start marketing planning early, ideally during product development.
- Align regulatory, clinical, and marketing teams.
- Invest in scalable systems to manage documentation and compliance.
- Continuously gather market feedback and adapt positioning.
Ultimately, the companies that succeed are those that treat marketing not as a final step, but as a strategic function embedded throughout the product lifecycle.
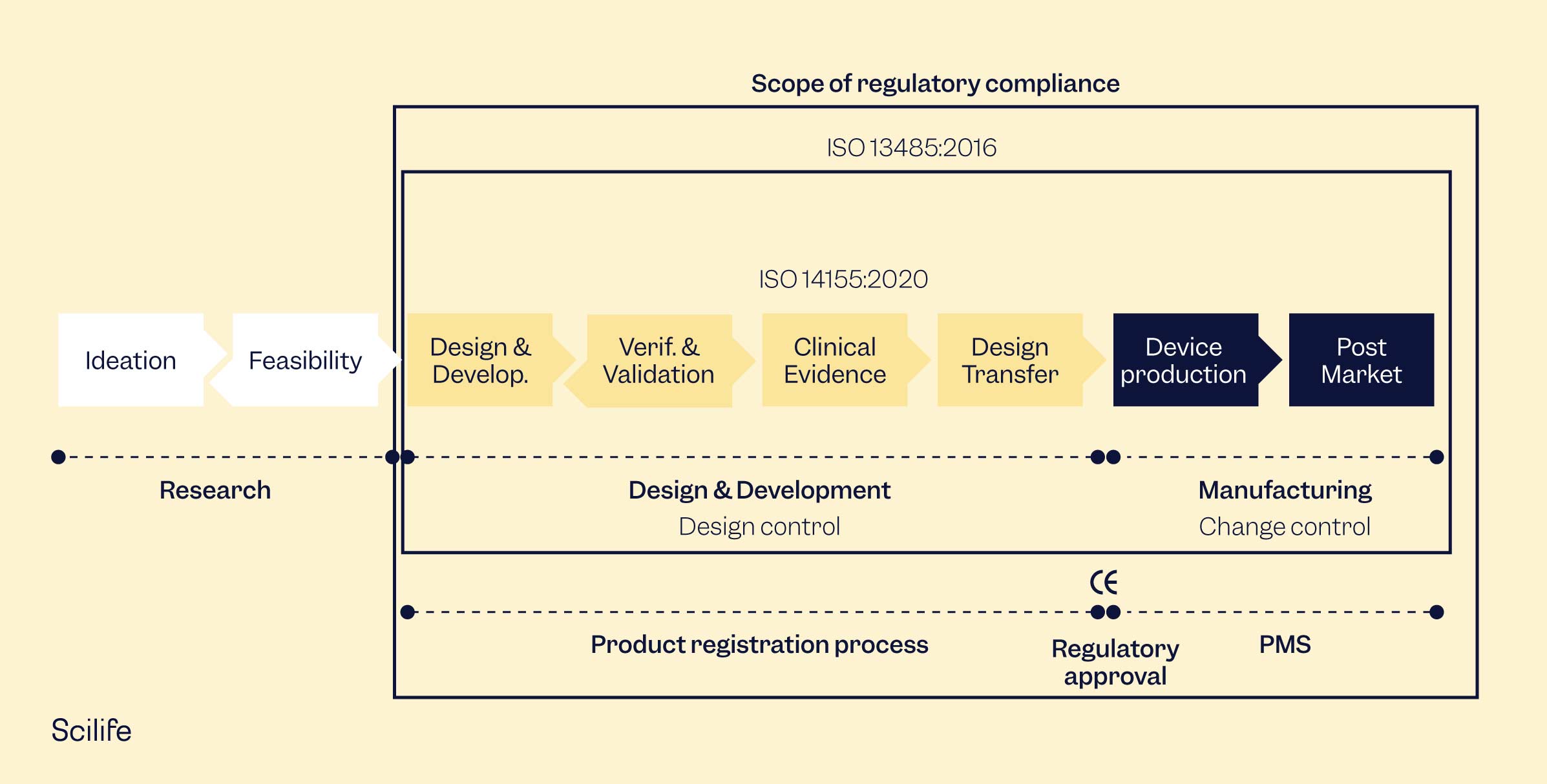
Device Development
Device development can be scary. It has many phases and requires thoughtful planning, execution, and revision. There are bound to be mistakes and surprises, and sometimes you want to rip your hair out. Once your device is ready to launch, though, it will all have been worth it. The pharmaceutical and medical device industries are some of the most heavily regulated. In every step of every process, there are documentation requirements and permissions needed. However, if you get organized early and are proactive about the requirements, you will find it is not as difficult as expected.
Design Controls
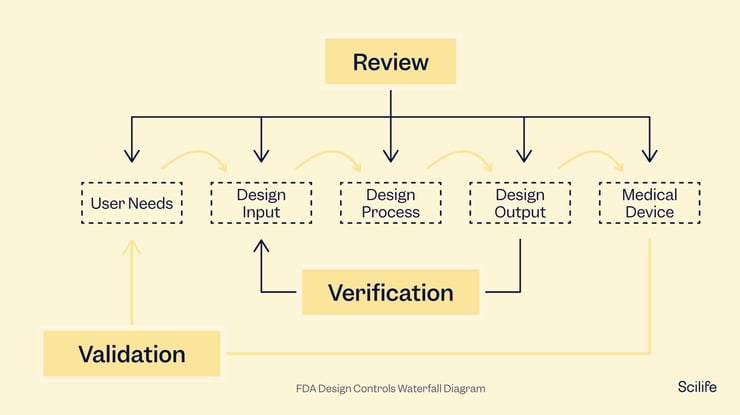
Design controls should be documented from the very beginning of device development and continuously through the entire process. If you only start when you are ready to launch your project, you are too late. Design controls can help you design a better product and avoid having to draft documentation for things you barely remember at the end of the project.
The FDA and ISO 13485 have similar documentation requirements for design controls, emphasizing the need for full traceability. You will need to keep track of all modifications made to the design along the way and document them. The most common method is through the traceability matrix, which any auditor or inspector will review to understand your device development process and the device itself. While some manufacturers create traceability matrices in Excel, considering a traceability matrix software could make your life much easier.
Design controls include design and development planning, user needs, design inputs and outputs, design verification and validation, design reviews, design changes, and the design history file. Each of these steps has its own requirements and should be guided by the standard being followed, whether the FDA or ISO 13485 or localized regulations.
Risk Management
The end goal of your medical device is to help patients and healthcare professionals. To do that, your device needs to be safe, and it should not put your users at unnecessary risk. Risk management is considering all the risks associated with using your medical device and mitigating them as far as possible.
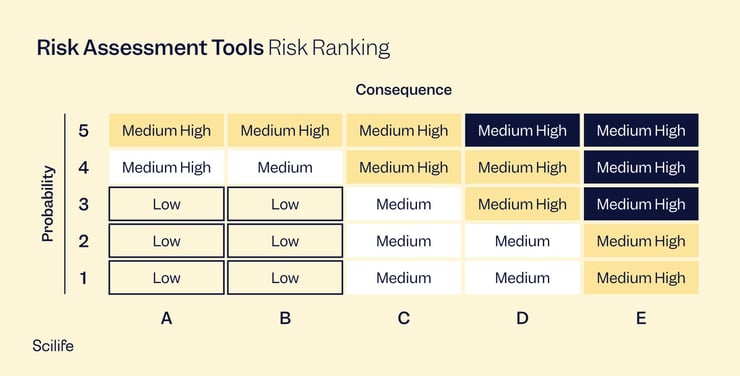
The gold standard for risk management is the international standard ISO 14971 Application of risk management to medical devices. While many regulatory people believe that failure mode and effect analysis (FMEA) is the same as ISO 14971, it is not the case. FMEA is a wonderful tool for risk management, but it is only a risk analysis process, not a complete system.
Risk management systems, according to ISO 14971, consist of a risk management plan, risk analysis, risk evaluation, risk controls, and an evaluation of overall risk acceptability. Risk management is a living thing that must be performed throughout your device's lifetime. The risk management process continues once your product goes to market. Production and post-production risk management activities, as well as any other post-production process that is in your QMS, should feed into your risk management process.
Quality Management System (QMS)
Your quality management system should be tailored to your company while complying with applicable regulations. Small companies don’t need a heavy QMS system, as their manufacturing and distribution chains will be small and easily manageable. In contrast, a larger company requires a different scale in its QMS system. For your quality system to pass regulatory scrutiny, there are some essential elements it should cover. Usually, document control and record management, design controls, risk management, and supplier management are vital elements in a simple QMS system. You will also need to consider post-market surveillance procedures, such as corrective and preventive action (CAPA) procedures, complaint handling, customer feedback, etc., once your device launch has been planned.
If you want to sell your device worldwide, ISO 13485 is the gold standard. If you sell only in the US, the 21 CFR Part 820 will suffice. The most important aspect of your QMS is that all your personnel know what they are supposed to do and actually do it. Having complicated procedures that no one follows is surprisingly common for manufacturers, and it is a problem you should solve sooner rather than later.
Technical Documentation
Technical documentation is required for your device, whether you are looking for market authorization in the EU, the US, or other regulatory markets. Generally speaking, technical documentation is a compilation of documents about a medical device and its development. It includes design, general device descriptions, intended use, and everything else required to comply with the applicable regulations. In the EU, the technical file is a key requirement for CE-marking, while the FDA requires the similar 510(k) for market authorization.
The technical documentation for a device should include sufficient information for the regulatory authority to thoroughly understand the device in question and its manufacturing process, as well as its compliance with local regulatory requirements (such as the General Safety and Performance Requirements (GSPRs) in the EU), benefit-risk analysis, risk management, verification and validation, post-market surveillance activities, and more.
The technical documentation typically has an expected format depending on the market.
Clinical Evaluation
Contrary to popular belief, clinical evaluation is one of the most important steps in preparing your device's technical documentation, along with risk management. The clinical evaluation is where you demonstrate that your device is safe and performs well when used as intended. It is where you summarize all the clinical evidence gathered and conclude whether the benefits of using your device outweigh the risks.
The clinical evaluation is based on clinical data gathered from various sources. For clinical evaluations drafted before a device goes to market, the primary clinical data will come from clinical trials, pre-clinical testing, and scientific literature. Not all devices will have clinical trials available, so for lower-risk devices, the clinical evaluation primarily consist of clinical literature.
Any clinical data you identify during the clinical evaluation process must be analyzed, appraised, and weighted in a clinical evaluation report. The clinical evaluation report summarizes all your clinical data and demonstrates that your device’s risk-benefit profile is acceptable.
Current guidance on clinical evaluations in the EU includes the EU MDR and MEDDEV 2.7/1 Rev 4. Although it is made for the European Union market, any clinical evaluation that complies with MEDDEV 2.7/1 will be accepted in all major regulatory markets.
Clinical evaluation should be performed before the device goes to market and continuously during the lifetime of the device.
Regulatory Submission
Now that you have all your documentation and device details in place, it is time to prepare the regulatory submission.
In the US, your regulatory submission will depend on your device classification. Class I devices typically do not require pre-market submissions as they are simple and low-risk. Class II devices, those classified as moderate risk, require 510(k) submission. The 510(k) is not approved but cleared – the 510(k) demonstrates equivalence with a device already on the market, so the 510(k) clearance only indicates that your device is equivalent to your predicate. It is not a device review and approval, per se. High-risk devices require PMA submission and approval before being placed on the market. The PMA undergoes thorough review by the FDA and typically requires clinical trials. Lastly, if your device is so innovative that no predicate exists on the market, the De Novo pathway is for you. De Novo devices are reviewed through a risk-based methodology and require robust risk mitigation. Market authorization takes between 30-180 days on average, depending on your device class.
In the EU, your device is subject to CE-marking, no matter the device classification. Even the lowest risk devices, Class I self-certified, must undergo conformity assessment, although, for these devices, third-party involvement by a notified body is not required. Class I, Class IIa and IIb, and Class III devices must undergo conformity assessment through a notified body that will review their technical documentation thoroughly. Devices cannot be put on the market unless granted CE-marking. Notified body review can take 12-18 months on average and longer if several feedback and revision rounds are required.
Device Launch
Your device is on the market – congratulations! The work doesn’t stop there, though. You could argue that the work has just begun. Market approval is just the beginning of the marathon, and many manufacturers are surprised at the amount of work still required once your device is on the market. Besides the logistics of producing medical devices, such as supply chain management, marketing material distribution, and manufacturing process, there are three crucial aspects to consider after your device launch.
Global Product Registrations
Most medical device manufacturers look to other markets once their target market has been conquered. Choosing which secondary markets to target is an exercise in itself, not to mention understanding the regulatory requirements of each one. Many manufacturers seek market authorization in regions adjacent to their target market – for example, many US manufacturers choose to expand into Latin America first. In contrast, manufacturers who obtained CE-marking in the EU and market their devices in Scandinavia might decide to move to Germany or the Netherlands next.
While the EU and the US are generally considered the “big markets” and frequently act as templates for the regulations in other countries or regions, individual differences exist between markets. Some might require complete pre-clinical testing reports, while others are alright with summaries. Some might require full technical files, while others are alright with only seeing the 510(k) clearance and a certificate of free sales/certificate of exportation. In most cases, hiring local consulting teams will be necessary for larger companies that do not have regulatory departments in the desired region and for smaller companies with offices only in their homeland, especially in cases where the local language is different. Regulatory requirements can seem simple, but they are usually anything but. A specialized local team is unavoidable for most geographies unless you have someone on staff who is already an expert in that market.
Post-Market Surveillance
Post-market surveillance is one of the most critical responsibilities after product launch. Post-market surveillance monitors the device’s behavior in the market and feeds that information back into the technical documentation to ensure a continuously safe device. It is a critical part of most regulations and plays a prominent role in the FDA and EU regulations.
While you may have tested your device’s safety in clinical trials or studies, you still need to understand how it behaves in a real-world setting. The FDA and the EU require post-market surveillance activities for regulatory compliance.
When talking about post-market surveillance, there are a few unavoidable activities. 1) submission of regular post-market surveillance reports to the relevant competent authority. These reports should summarize the adverse events that occurred in the previous period, providing an overview of how the device behaves under real-world use. 2) vigilance procedures establishing processes for reporting adverse events to the authorities. 3) post-market clinical follow-up to evaluate and manage any reported risks, failures, non-conformities, or other critical safety issues related to using the device. Post-market clinical follow-up is a significant part of the updated MDR and hence a critical part of EU MDR compliance. 4) trend reporting to determine if any new risks are emerging or if a particular safety issue is causing frequent non-reportable events. 5) complaint management to determine how your users are experiencing the device. 6) corrective and preventive action (CAPA) management to monitor and correct any quality or safety issues during device use.
Updates to Technical File
Your EU technical file will require frequent updates. Depending on the risk class of your device and the document in question, you are looking at updating documentation every 1-5 years. The technical file should truly be a living document, subject to modifications throughout the lifetime of your device.
You might be wondering when exactly you should be updating your technical file. The short answer is every time you receive information that changes the benefit-risk profile of your device or otherwise modifies how your device meets the general safety and performance requirements.

Discover how a Smart Quality Management System (QMS) can help you easily comply with GMP standards





