Device Master Record (DMR)
What is the Device Master Record?
The Device Master Record (DMR) is a compilation of records needed to produce a medical device. Whenever you are planning to manufacture a new medical device, you need to generate and record all necessary information on the new device – including design requirements, specifications for production, quality assurance (QA), packaging, labeling specifications, and more.
Some records needed for the DMR may remind you of what was required to generate the Design History File (DHF) such as device specifications, packaging, and labeling specifications. And that is correct! As the DHF was created at the beginning of your design process, it already includes parts of the design outputs. And if you have prepared a well-documented and -organized DHF, you can easily reuse some documents for your DMR.
What Are the Legal Requirements of a Device Master Record?
Both US and EU regulations mandate medical device manufacturers to comply with specific requirements for the design of medical devices.
For US regulations, the FDA 21 CFR defines DMR in subchapter “H - Medical Devices, Part 820 - Quality System Regulation, Subpart M - Records,” section 820.181 as follows:
820.181 Device master record.
Each manufacturer shall maintain device master records (DMRs). Each manufacturer shall ensure that each DMR is prepared and approved in accordance with § 820.40. The DMR for each type of device shall include, or refer to the location of, the following information:
(a) Device specifications including appropriate drawings, composition, formulation, component specifications, and software specifications;
(b) Production process specifications, including the appropriate equipment specifications, production methods, production procedures, and production environment specifications;
(c) Quality assurance procedures and specifications, including acceptance criteria and the quality assurance equipment to be used;
(d) Packaging and labeling specifications, including methods and processes used; and
(e) Installation, maintenance, and servicing procedures and methods.
Although European legislation does not provide specific definitions for DMR and DHF, the ISO 13485:2016 standard on “Medical devices – quality management systems – requirements for regulatory purposes” states that all documentation shall be contained in a single file referred to as the “Medical Device File” (see section 4.2.3).
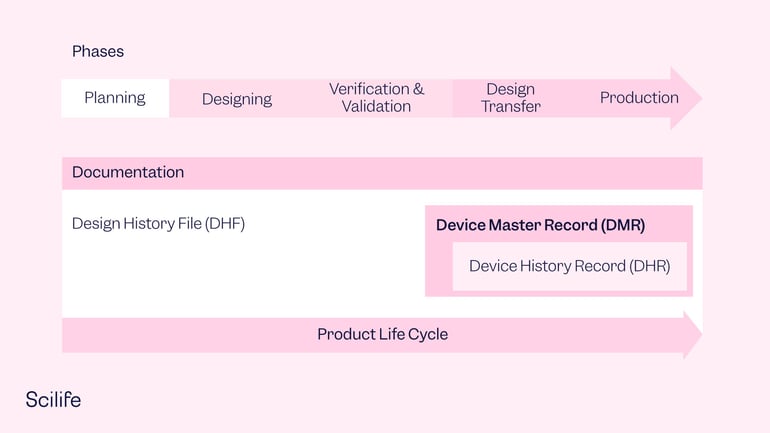
Elements of the Device Master Record
The DMR should include everything from the beginning to the end of the production and shipping process, as well as any applicable installation instructions. DMR documents can vary, including device specifications, compositions, drawings, formulations, software specifications, process specifications, appropriate equipment, methods, procedures, and environments, including cleanrooms, humidity controls, QA procedures, and any specialized equipment needed.
Ensuring that you have correctly generated, labeled, and archived the necessary documentation of each process step for the DMR, will make your work not only much easier today, but also for future audits. However, it can be even easier if you first establish a documentation management system in a digital environment. A well-established quality management system provides multiple benefits as well as control and easy maintenance of your documents and records. The DMR contains different records from different process steps, but that doesn’t mean you should duplicate those records to create a DMR folder for your medical device. If you do so, you will need an extra binder or two!
Once you start generating the required documents, you will realize that most of the documents you have created so far can be referenced in DMR. A well-established document management system within your quality management system can provide document control features such as document search, version control, access controls, updating, linking, and training. If you do not have a software system in place for this, it will be extremely difficult to stay well-organized and up-to-date with your device master records.
To clarify a little bit the differences between DMR, DHR and DHF concepts, the DHF should define how to design and develop your medical device; but the DMR contains all the required information to manufacture the finished product. Section 820.181 of the FDA QSR lists the key elements that should be included in the DMR:
1. Device specifications
2. Production process specifications
3. QA procedures and specifications
4. Packaging and labeling specifications
5. Installation, maintenance, and servicing procedures and methods.
Here are a few DMR examples that would be required to manufacture the product:
- Manufacturing test documents
- Process flow checklist
- Work instructions
- Mixing instructions
- Sterilization instructions
- Assembly instructions
- Packaging instructions
- Design directive
- Service manual
- Inspection procedure
- Parts lists
- Bill of materials (BOM)
- Labeling
- Device label
- Packaging label
- User instructions
- User guide
- Quick start guide
- Safety manual
- Purchasing specifications
- Drawings
- Product, plastic components, primer, and seconder packaging
- Component, packaging, and software specifications
- Composition formulation
- Schematic diagrams
- QA procedures and specifications
- Acceptance criteria and QA equipment to be used
Additional resources

How to Implement the Continuous Improvement Cycle | Scilife
Even an organization with stellar leadership and a solid core of employees experiences hiccups from time to time. Despite having assembled all the ...
How to assess and enhance your Quality Management Maturity | Scilife
As the life sciences industry becomes increasingly regulated and competitive, quality management has become more vital than ever. Are you confident ...

8 Best QMS Software for Life Sciences (2026 Comparison)
The right electronic Quality Management System (eQMS) can help strengthen your compliance processes and build a culture of quality within your ...

How to write a good quality plan for medical devices | Scilife
In life sciences, especially if you’re in the medical device industry it becomes harder to manage projects in accordance with your company’s quality ...
Turn quality into your brightest asset with Scilife

