Device History Record (DHR)
A Device History Record (DHR) includes everything you need to manufacture the medical device. The history and data of how you manufacture the medical device according to the DMR are stored in the DHR. Similarly, the DHFis the history of the design; the DHR is the history of the device
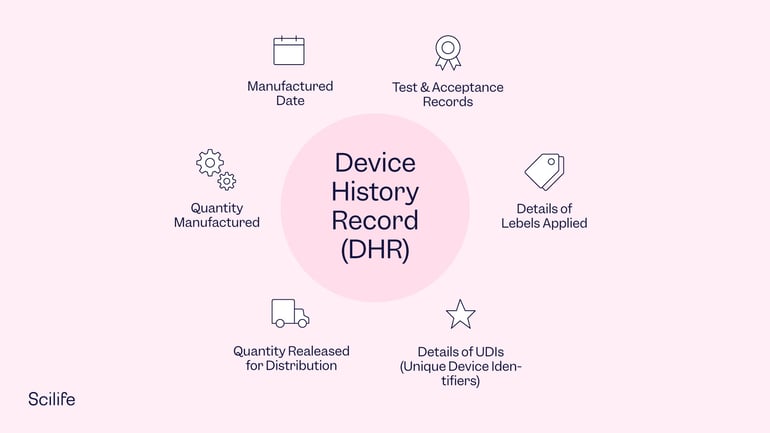
The DHR includes records of the manufactured products and helps to ensure every product that you produce is on track according to the DMRs. A normal DHR should include the following documents per FDA Part 820.184 Device History Record.The FDA states that:
Each manufacturer shall maintain device history records (DHRs). Each manufacturer shall establish and maintain procedures to ensure that DHR's for each batch, lot, or unit are maintained to demonstrate that the device is manufactured in accordance with the DMR and the requirements of this part. The DHR shall include, or refer to the location of, the following information:
(a) The dates of manufacture;
(b) The quantity manufactured;
(c) The quantity released for distribution;
(d) The acceptance records which demonstrate the device is manufactured in accordance with the DMR;
(e) The primary identification label and labeling used for each production unit; and
(f) Any unique device identifier (UDI) or universal product code (UPC), and any other device identification(s) and control number(s) used.
However, ISO 13485 has no specific section for the DHR term.Section 7.1 - Planning Product Realization states that the organization should define product realization steps and ensure that it is compatible with the requirements of other QMS> (Quality Management System) processes. The organization should specify the following where applicable:
- Product quality objectives and requirements
- Defining processes and providing documents and resources specifically for the product, including infrastructure and a work environment>
- Required traceability activities of all processes specific to the product, together with the criteria for product acceptance
- Documents are required to prove that the realization process and resulting product meet external requirements
Medical device QMS software makes it easy to maintain the structure of a DHF.
Why is the Device History Record so important?
The DHR is the most crucial part of your product. You should create a DHR file and maintain it during the entire product shelf life – or longer, depending on your internal or external requirements. DHR is not only a requirement that you need to comply with, it is also your lifesaver if any issues do arise. A well-documented DHR can provide evidence of when and what you did, as well as of how the product was manufactured, controlled, and approved according to predefined and validated specifications before receiving it from a customer. As you start to understand the grave importance of the DHR, you will soon realize that you need to improve your product realization process and DHR content.
DHRs are a crucial tool during audits, as typically auditors pick a specific lot from your previously manufactured products and review all related activities, preferably within your DHR documentation to see whether your product remained compliant with external requirements, internal procedures, and DMR (Device Master Record) . The DHR documentation allows you to show traces of the performed activities from manufacturing to the end of product realization.
Furthermore, DHR is also a vital building block during customer complaint activities. To visualize it, imagine that you received a customer complaint about a specific lot of your medical device. The DHR that belongs to this particular UDI (Unique Device Identification System) enables you to access all details regarding when it was produced, which QC tests were performed, what the results of these tests were, and much more.
After receiving a customer complaint, you must conduct an investigation and root cause analysis. You should also review all historical data regarding the medical device and/or complaint. The output can enable you to decide whether this was a one-time event or a recurring problem leading to the issue of a CAPA (Corrective and Preventive Action) report to initiate an extensive investigation.
To investigate a complaint that belongs to a specific ID, follow these steps:
Perform a gap analysis
Here are the most common gaps identified during a gap analysis.
- Batch release activities without QA involvement
- Missing and/or inadequate documentation
- Failure to comply with specifications
- Lack of traceability
- Process failures due to missing control points for critical parameters
Meet with employees who need to be involved in the Device History Record,
Meeting with representatives of all involved departments is essential since the DHR is a compilation of your multidisciplinary processes. You should assess who needs to be a part of your DHR, so that you can compile comprehensive DHRs without missing out on data. Typically, the involved departments are (at minimum) as follows:
- Manufacturing
- Packaging
- Distribution
- Record Control
- Quality Control
- Quality Assurance
- Regulations
- Engineering
- Marketing
Double-check existing Device History Records
The DHR should cover all details and tracks that belong to the specific product. Double-checking DHR documents ensures that no document is missing, no data were left out, and all results meet the acceptance criteria.
Determine all critical parameters
The overall manufacturing process should be analyzed as part of the compilation for the DHR release process. Once the process is in place, determine the critical parameters or control points.
Additional resources

How to Implement the Continuous Improvement Cycle | Scilife
Even an organization with stellar leadership and a solid core of employees experiences hiccups from time to time. Despite having assembled all the ...
How to assess and enhance your Quality Management Maturity | Scilife
As the life sciences industry becomes increasingly regulated and competitive, quality management has become more vital than ever. Are you confident ...

8 Best QMS Software for Life Sciences (2026 Comparison)
The right electronic Quality Management System (eQMS) can help strengthen your compliance processes and build a culture of quality within your ...

How to write a good quality plan for medical devices | Scilife
In life sciences, especially if you’re in the medical device industry it becomes harder to manage projects in accordance with your company’s quality ...
Turn quality into your brightest asset with Scilife

