

Life science industries have their language and it consists of many concepts, terms, and abbreviations that are commonly mixed up. When you see them together it is even harder to grasp the nuances between concepts.
Today we’ll give you a long list of concepts with their definitions and small differences among others. Keep reading and also bookmark this page to come back again when you feel lost between quality concepts.
Unveiling General Quality Concepts
In this section, you will learn general quality concepts that every life science industry knows and uses.
If you want to learn more specific concepts with their definition and differences, keep reading! We also included a list of industry-specific concepts in the following sections.
Audit vs. Inspection
Audit
A verification activity, such as an inspection or examination, of a process, product, or quality system to ensure compliance with requirements. It could be conducted on-site, or virtually. An audit could apply to an entire organization or be specific to a function, process, production step, or product. Audits are systematic, independent, and documented processes to collect evidence(s) and evaluate it objectively as per audit criteria.
Inspection
A verification activity that involves measuring, examining, testing, and gauging one or more characteristics of a product or service. Its purpose is to determine whether each characteristic conforms to specified requirements.
| Criteria | Audits | Inspections |
| Purpose | are generally conducted to assess the effectiveness of a QMS. | are focused on compliance with specific regulations. |
| Who conducts | can be internal (conducted by the organization itself) or external (undertaken by third parties). | are usually undertaken by regulatory authorities. |
| Scope | can be broad and cover multiple aspects of quality management. | are often more focused and specific. |
| Frequency | are usually scheduled and periodic. | can be unannounced, and may be triggered by specific events or complaints. |
| Outcome | The outcome of an audit is generally a set of recommendations for improvement. | The outcome of an inspection can lead to regulatory actions, including fines or sanctions. |
Calibration vs. Qualification vs. Validation vs. Verification
Calibration
The set of operations that establish, under specified conditions, the relationship between values indicated by a measuring instrument or measuring system, or values represented by a material measure, and the corresponding known values of a reference standard. (“EU GMP Glossary”)
Qualification
The action of proving that equipment works correctly and leads to the desired results. It is often considered a part of a validation. (“EU GMP Glossary”)
Validation
The action of proving that a procedure, process, system, or equipment works correctly and leads to the expected results. It is sometimes widened to incorporate the concept of qualification. (“EU GMP Glossary”)
Verification
The process of confirming a product, service, or system meets specified criteria or standards. It is the act of determining whether products and services meet specific requirements This evaluation is carried out by providing objective evidence that confirms the fulfillment of specified requirements.
| Calibration vs. Validation | Calibration vs. Qualification | Validation vs. Qualification |
| Calibration is specific to measuring instruments & systems to ensure their accuracy against certified/known standards. Validation ensures that a piece of equipment, process, or system will do what it is supposed to do. | Qualifying equipment or systems is accomplished through calibration. Calibration is concerned with the accuracy of measuring instruments. Qualification is concerned with the suitability of the equipment or system for its intended use. | The term validation is more comprehensive, which may include qualification as well. Validation ensures that a system, as a whole, meets the desired objectives, while qualification would be a part of that system, focusing on individual pieces of equipment. |
CAPA vs. RCA
CAPA
A quality process to identify, analyze, and investigate the information, and take appropriate and effective corrective and/or preventive action to reduce and or eliminate product and quality problems or issues of a specific process.
RCA
A problem-solving analysis technique to identify the underlying cause of a problem or incident, solve it, and prevent recurrence. Uncovering the correct and accurate reason(s) why the problem or issue is happening or has already occurred. It is often a step within the CAPA process to determine what went wrong so that corrective actions can be taken.
|
Critera |
CAPA | RCA |
|
Scope |
is broader in scope and includes both corrective and preventive actions. | specifically focused on identifying root causes. |
| Application | RCA is often a component of CAPA. After RCA identifies the root cause, CAPA is used to implement corrective and preventive measures. | |
| Objective | prevent recurrence and occurrence of nonconformities. | identify the underlying reason for a specific problem. |
Change Control Management vs. Configuration Management
Change Control Management
Change Control Management involves the process of managing, documenting, planning, validating, and implementing the change and verifying the effectiveness of change within a quality management system to ensure consistency and compliance. It's closely related to CAPA as described in FDA's CFR 21 Part 820, which outlines procedures for analyzing processes, investigating causes of nonconformities, and implementing necessary changes to prevent recurrence.
Configuration Management
A practice of systematically managing and maintaining system and product information, versions, and requirements. It ensures that the current design and build state of the system is known, good, trusted, and recorded.
|
Critera |
Change Control Management | Configuration Management |
|
Focus |
change process and its impact. | on the item being changed and its state. |
| Scope | often has a broader scope, affecting multiple parts of an organization. | might be limited to IT or specific systems. |
| Regulatory Compliance | often more tightly linked to regulatory compliance due to the critical nature of the changes being made. | |
| Risk Management | involves more extensive risk assessment compared to Configuration Management. | |
| Documentation | often requires more detailed documentation, including justifications for the change, impact assessments, and approvals. | requires documentation. |
Checklist vs. Checksheet
Checklist
A tool for ensuring all important steps or actions in an operation have been taken. Checklists contain items important or relevant to an issue or situation.
Check sheet
A simple data recording device to collect and analyze data as a record of events, occurrences, or frequency of specific factors. The check sheet is custom-designed, which allows you to readily interpret the results.
|
Critera |
Checklist | Check Sheet |
|
Purpose |
Ensures that a set of tasks or criteria are completed or verified. | It’s mostly used for data analysis and data collection. |
| Structure | Usually a simple list of items that need to be checked off. | Often has a more complex structure, designed to collect specific types of data. |
| Usage | Used for the preparation or verification processes. | Used for observational studies, inspections, or QC processes. |
| Ability to adapt | Generally rigid and follows a set structure. | It can be more flexible and tailored to the specific data being collected. |
| Data analysis | Not designed for data analysis. | Specifically designed to facilitate data analysis. |
CoQ vs. CoPQ
Cost of Quality (COQ)
COQ refers to the total cost of activities related to the quality of the product or service, including prevention, appraisal, and correction of defects in products or services.
Cost of Poor Quality (CoPQ)
COPQ refers to the costs of activities associated with providing poor quality (defective) products or services such as waste, rework, returns, lost sales, and failures.
|
Critera |
Cost of Quality (COQ) | Cost of Poor Quality (CoPQ) |
|
Scope |
includes all costs related to the quality, including prevention and appraisal. | focuses only on the costs of defects. |
| Objective | aims to optimize total quality costs. | aims to reduce defect-related costs. |
| Components |
include prevention and appraisal costs. |
These are not part of CoPQ. |
Defect vs. Defective
Defect
Refers to a specific issue or flaw in a product or process that deviates from the intended design or functionality. (“EU GMP Glossary”)
It is a nonfulfillment of a product or service for its intended requirement or reasonable expectation for use, including safety considerations. A defect could be a non-conformance of a possible quality characteristic of a unit which could cause customer dissatisfaction.
Classes of defects

Defective
Refers to a product or process that contains one or more defects, making it unfit for its intended use. A defective unit means a unit of product that contains one or more defects concerning the quality characteristic(s) under consideration.
|
Critera |
Defect | Defective |
|
Scope |
refers to a specific issue, and defective describes it. | refers to the overall condition of a product or process containing defects. |
| Implication |
is a characteristic. |
is a classification. |
|
A product with a defect is not necessarily defective if the defect doesn't affect its overall functionality. |
||
| Usage |
is often used in quality control to identify specific issues for correction. |
is used to describe products that may need to be recalled or discarded. |
Deviation vs. Nonconformity
Deviation
Refers to a departure from the originally specified, approved requirements or established standard of a product or a service before its realization. Deviations can be either planned or unplanned and may require corrective action(s).
Nonconformity
Refers to the failure to fulfill a requirement, intended state, or specification.
|
Critera |
Deviation | Nonconformity |
|
Scope |
Usually relates to processes and procedures. |
Relates to products, processes, systems, or the quality management system itself. |
| Timeframe |
Usually temporary and may be planned or unplanned. |
is usually identified after the fact during inspections or audits. |
| Action Required |
Deviation may require immediate corrective action. |
usually triggers a formal corrective and preventive action (CAPA) process. |
| Documentation |
often requires immediate documentation for traceability. |
is usually documented as part of a quality audit or inspection. |
| Regulatory Impact |
may or may not have a regulatory impact depending on its nature. |
In many cases, it has a direct regulatory impact and can result in non-compliance penalties. |
Different types of audits (ISO 9001:2015)
Audit
Verification activities, such as an inspection or examination, are performed on a process, product, or quality system to ensure compliance with requirements. Audits can be conducted on-site or virtually. They could be conducted by an organization as a whole or by a specific function, process, process step, or product. An audit is a systematic, independent, and documented process that collects evidence(s) and evaluates it objectively by the audit criteria.
|
Critera |
Combined Audit | External Audit | Internal Audit | Process Audit | Product Audit |
|
Purpose |
Aims for combined standard improvements by sharing audit costs, time, and resources. |
Compliance with external standards. |
Improve and enhance organizational processes and controls for continuous organizational improvement. |
Continuous process improvement of specific processes and defect prevention. |
Ensures products meet quality and safety requirements and desired specifications. |
| Expertise |
✓ Requires auditors knowledgeable in multiple standards. |
✗ Specific to one standard or regulation. |
✓ In-depth knowledge of internal systems. |
✓ In-depth knowledge of specific processes. |
✓ In-depth knowledge of product specifications. |
| Coordination |
✓ between multiple organizations. |
✗ No need for external coordination. |
✗ Internally. |
✗ Internally. |
✗ Internally. |
| Independence |
✗ Depends on the involved organizations. |
✓ Conducted by an independent entity. |
✗ Dependent on the organization's internal team. |
✗ Dependent on the organization's internal team. |
✗ Dependent on the organization's internal team. |
| Certification |
combined certification. |
certification or assessment. |
doesn't lead to certification. |
doesn't lead to certification. |
May lead to product certification or rejection. |
| Regulatory Compliance |
✓ Ensures compliance by combining standards with a single audit. |
✓ Company compliance is assessed with external standards. |
✓ Helping to comply with internal standards and regulations. |
✓ Assesses effectiveness, compliance, and outputs. |
✓ Verifies against product regulations, standards, attributes, and compliance. |
Compliance vs. Conformance
Compliance
An organization's ability to meet specific requirements, contract terms, regulations, or standards within an organization.
Conformance
A statement indicating a product’s or service’s compliance with specified requirements, contracts, or regulations.
Conformity Assessment vs. Declaration of Conformity
Conformity assessment
A set of activities concerned with determining whether relevant requirements in standards or regulations are met, such as sampling, testing, inspection, certification, management system assessment and registration, accreditation of competence for those activities, and recognition of the capabilities of accreditation programs.
Declaration of Conformity
Refers to a formal declaration from the manufacturer or supplier stating that a product, service, or system complies with specific standards or regulations. It is based on the results of conformity assessments and is usually accompanied by supporting documentation.
Correction vs. Corrective Action vs. Preventive Action
Correction
Refers to an immediate action that is taken to address a detected nonconformity. It deals directly with the symptom of the problem, providing an immediate solution, but does not necessarily address the root cause of the problem.
Corrective action
A corrective action is taken in response to a nonconformity. It’s a set of steps to identify, address, and reduce or eliminate the root cause(s) of a nonconformity to prevent its recurrence. Unlike corrections, corrective action targets the underlying issue and root cause.
Preventive action
As opposed to corrective actions, preventive actions are taken to prevent potential nonconformities or problems before they occur by making predictions, trend analyses, or risk assessments.
|
Critera |
Correction | Corrective Action | Preventive Action |
|
Purpose |
Address the detected issue immediately. |
Eliminate the root cause of a problem. |
Prevent potential problems from occurring. |
| Approach |
Reactive |
Reactive |
Proactive |
| Basis |
Based on the symptoms of the problem. |
Based on the root cause analysis. |
Based on the predictions, trend analyses, or risk assessments. |
| Independence |
Short-term. |
Long-term after the problem occurs. |
Long-term solution before the problem occurs. |
| Duration of the solution |
combined certification. |
certification or assessment. |
doesn't lead to certification. |
| Outcome |
Immediate containment of the issue. |
Prevention of issue recurrence. |
Prevention of issue emergence. |
CSV vs. CSA
CSV (Computer System Validation)
It is a documented process to ensure that a computerized system produces consistent and reliable results in line with predetermined specifications, requirements, and its intended purpose.
CSA (Computer Software Assurance)
This approach emphasizes cybersecurity, software lifecycle processes, and post-market surveillance, focusing on risk management, patient safety, and assurance activities.
With CSA, documentation-heavy processes are shifted to critical thinking and assurance activities.
| CSV | CSA |
|
Planning |
Identifying the intended use |
|
Defining DS, FS, and URS |
Determining risk-based approach |
|
Testing includes: Verifying IQ against DS Verifying FS against PORQUE Verifying URS against OQ |
Determining appropriate assurance activities |
|
Reporting all test-based evidence |
Establishing appropriate records |
Document Control vs. Document Management
Document Control
A process to ensure that documents are reviewed, updated, approved, and obsoleted properly so that documents remain consistent and compliant.
Document Management
Compared to document control, it’s a broader system that covers the end-to-end process of handling documents, including creation, storage, retrieval, sharing, tracking, revision, and archiving.
GDP vs. GDocP
| Criteria | Good Distribution Practices (GDP) | Good Documentation Practices (GDocP) |
|
Definition |
Guidelines for the proper distribution of medicinal products for human use. |
Guidelines for recording, correcting, and managing documents, particularly in regulated industries. |
|
Focus |
Ensuring the quality and integrity of drugs throughout the supply chain. |
Ensuring clarity, accuracy, and integrity in documentation. |
|
Application |
Wholesalers, carriers, and everyone involved in the chain from manufacturer to end user. |
Anyone involved in the creation, handling, or review of documentation. |
|
Objective |
To prevent contamination, and mix-ups, and to ensure product quality during distribution. |
To create a reliable and consistent record that can withstand scrutiny, especially in audits. |
GxP vs. GMP
GxP
The general term for guidelines and regulations for good (anything) practices. You can replace the "x" with various fields such as "Manufacturing (M)", "Clinical (C)", and "Laboratory (L)". Many regulated sectors rely on GxP to ensure quality and consistency.
| G | x | P |
| Good | Variables | Practices |
Good Manufacturing Practices (GMP)
Most known term for a set of practices recommended or required by the regulatory agencies for manufacturers to ensure their products consistently meet requirements for their intended use. The GMP covers all aspects of production, starting from raw materials and facilities totraining and personal hygiene.
Innovation vs. Invention
| Criteria | Innovation | Invention |
|
Definition |
The implementation of a new or improved product, process, or service. |
The creation of a novel idea or method that could lead to a new product, process, or service. |
|
Focus |
The implementation and commercialization of a new or improved product, process, or service that provides value and meets specific market needs. |
The creation of new ideas or methods. |
|
Application |
Broadly adopted in the market. |
There may not be a commercialization or adoption in the market yet. |
Instrument vs. Equipment
| Criteria | Instrument | Equipment |
|
Definition |
|
|
|
Scope |
Used for specific, often precise tasks. |
Used for general purposes. |
|
Complexity |
Can be part of a larger equipment. |
Can encompass multiple instruments. |
IQ vs. OQ vs. PQ
| Criteria | IQ (Installation Qualification) | OQ (Operational Qualification) | PQ (Performance Qualification) |
|
Definition |
Confirms an equipment or system has been installed correctly according to specified requirements. |
Confirms an equipment or system operates as intended across all specified operating ranges. |
Confirms an equipment or system consistently performs according to the intended use in actual production conditions. |
Out of Specification (OOS) vs. Out of Trend (OOT) vs. Out of Expectation (OOE)
| Criteria | OOS (Out of Specification) | OOT (Out of Trend) | OOE (Out of Expectation) |
|
Definition |
Outside of acceptance criteria. |
Consistent and significant trends that are away from expected conditions over time. |
Specified limits are unexpected, or deviate from historical data or patterns. |
Process Control vs. Process Capability
| Criteria | Process Control | Process Capability |
|
Definition |
Methods used to control, manage, and maintain a process, ensuring it operates at its full potential. |
A statistical measure of a process's ability to produce within specification limits. |
|
Objective |
Ensuring that the process consistently operates and meets expected specifications. |
Determining if the process inherently meets expected specifications. |
|
Focus |
On the real-time monitoring and adjustments. |
On the analysis of historical data to determine potential future performance. |
|
Methodology |
Can use feedback loops, control charts, alarms, etc. |
Uses statistical measures like Cp, and Cpk to evaluate capability. |
|
Application |
Continuous during the operation of the process. |
Often used during process setup or during reviews to ensure process suitability. |
QA vs. QC
Quality Assurance
Refers to all the planned and systematic activities implemented within the quality system that can be demonstrated to provide confidence that a product or service will fulfill requirements for quality.
Quality Control
It refers to the operational techniques and activities used to fulfill requirements for quality. Often, however, “quality assurance” and “quality control” are used interchangeably, referring to the actions performed to ensure the quality of a product, service, or process.
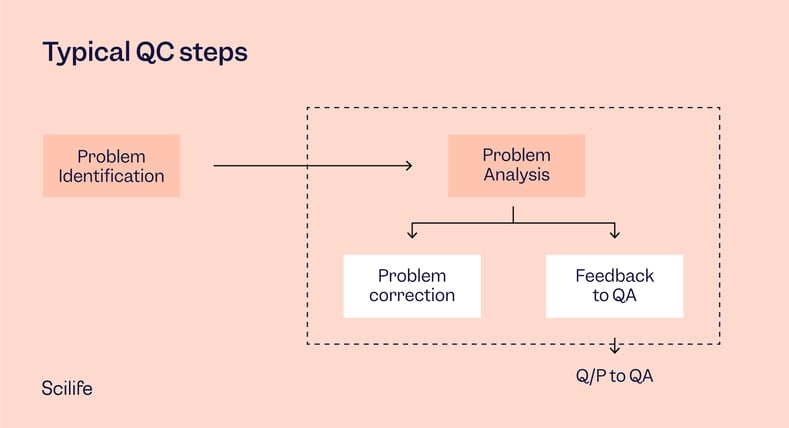
Further reading - Quality Assurance vs. Quality Control: Top 5 Differences
QMS vs. TQM
QMS (Quality Management System)
A formal system that documents the structure, processes, roles, responsibilities, and procedures required to achieve quality objectives.
Total Quality Management (TQM)
A management philosophy focused on embedding a culture of continuous improvement, customer satisfaction, and quality in all organizational processes.
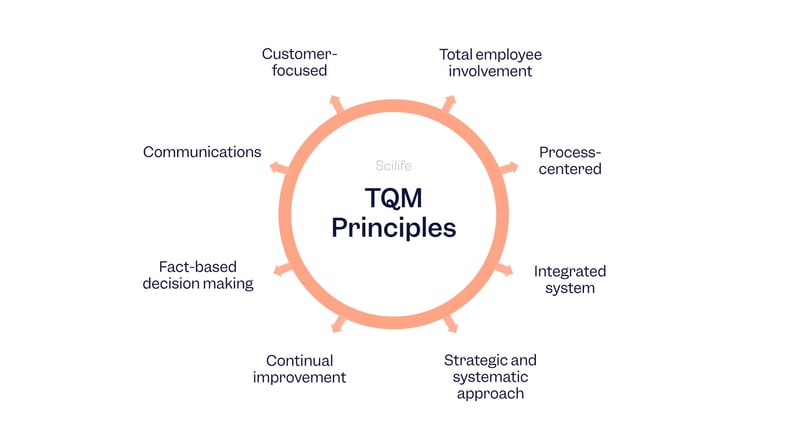
Quality tools for supplier audits and corrective action plans
Quality tools used to manage supplier audits and corrective action plans are a core part of an effective quality management system. These tools provide structured methods for qualifying suppliers, assessing audit risk, documenting nonconformances, and managing corrective and preventive actions over time.
By integrating supplier audit activities with CAPA processes, quality teams can ensure that identified issues are systematically investigated, addressed, and verified for effectiveness. This alignment supports continuous improvement, strengthens supplier quality oversight, and helps organizations meet regulatory expectations for risk-based quality management.
Risk Management vs. Risk Assessment vs. Hazard Analysis
Risk Management
Involves identifying, evaluating, controlling, monitoring and prioritizing risks with the aim of reducing their likelihood, mitigating their impact, or capitalizing on opportunities.
Risk Assessment
The process of identifying potential risks, estimating their impact, and determining their likelihood. It refers to the overall process and contains risk analysis and risk evaluation.
Hazard Analysis and Critical Control Point (HACCP)
Identification of potential hazards and assessment of the potential impacts of those hazards. FDA and USDA (Department of Agriculture) establish HACCP regulations for various sectors.
SOP vs. WI
Standard Operating Procedure (SOP)
It is a set of written instructions that outlines the overall aim and the step-by-step process to perform a specific task or procedure.
Work Instructions (WI)
It is a detailed breakdown of specific actions or steps that are required to complete a particular task, often a subset or more detailed component of an SOP.
| Criteria | SOP | WI |
|
Definition |
Instructions for a specific task or procedure. |
Detailed steps for a specific task. |
|
Depth |
General or overarching procedures. |
More detailed, often a subset of an SOP. |
Quality by Design vs. Quality by Testing
| Criteria | Quality by Design | Quality by Testing |
|
Definition |
Building quality from the outset. |
Making sure the finished product is of high quality by testing it. |
|
Approach |
A systematic approach to product development that emphasizes building quality into the product from the outset through design and development |
An approach where quality is ascertained by testing finished products to ensure they meet specified criteria. |
|
Focus |
On proactive design and development. |
Reactive, it is based on testing outcomes. |
Industry-Specific Quality Concepts:
Pharmaceutical Industry
Aseptic vs. Sterile
Aseptic
Refers to the prevention of contamination by harmful microorganisms. This technique ensures an environment (aka aseptic area) remains free from pathogenic contaminants, often through the use of sanitized equipment and controlled environments.
Sterile
Describes a condition where all living microorganisms have been destroyed or eliminated. A sterile environment or product is completely devoid of life, including spores and viruses.
| Criteria | Aseptic | Sterile |
|
Definition |
Prevention of contamination by harmful microorganisms. |
Complete elimination or destruction of all microorganisms, including spores and viruses. |
|
Objective |
To maintain an environment free from contamination. |
To ensure that an item or environment is entirely free of microbial life. |
|
Application |
Often used in pharmaceutical filling processes, surgery environments, etc. |
Used in autoclaving, radiation, and other sterilization methods. |
|
Extent |
Does not guarantee the absence of microorganisms, but minimizes their presence. |
Guarantees the total absence of microorganisms. |
Batch Release vs. Product Release
Batch Release
It refers to the approval of a manufactured batch after it has met the required quality tests and criteria. The entire batch is evaluated based on set standards and is released for distribution only after approval. One batch can contain 100 units or 100.000 units.
Product Release
It refers to the authorization for a final product to be sold or supplied to the end-user or the next stage in the supply chain. This is usually after it has been packaged and labeled.
| Criteria | Batch Release | Product Release |
|
Definition |
Approval of a manufactured batch after meeting required tests and criteria. |
Authorization for a final, packaged, and labeled product to be sold or distributed. |
|
Stage |
Occurs after batch production and before packaging. |
Occurs after the product has been packaged and is ready for the market. |
|
Objective |
Ensure the batch meets all quality standards before moving to the next production step. |
Ensure the final product is compliant and safe for the consumer or the next supply stage. |
|
Scope |
Refers to a specific batch or lot of products. |
Can apply to individual items within a batch or the entirety of products in a batch. |
CAPA vs. Deviation
Corrective and Preventive Action
CAPA is a quality process to identify, analyze, and investigate information, and take appropriate and effective corrective and/or preventive action to correct nonconformities, reduce and/or eliminate product and quality problems, and prevent their recurrence within a specific process.
Deviation
Refers to a departure from the originally specified, approved requirements or established standard of a product or a service before its realization. Deviations can be either planned or unplanned and may require corrective action(s).
| Criteria | CAPA | Deviation |
|
Definition |
A systematic approach to address and prevent nonconformities. |
A departure from standard procedures or specifications. |
|
Objective |
To identify, rectify, and prevent nonconformities from reoccurring. |
To document any unexpected events that depart from a standard or procedure. |
|
Scope |
Encompasses both corrective actions (addressing the issue) and preventive actions (ensuring it doesn't recur). |
Primarily focused on the identification and documentation of the deviation. |
|
Outcome |
Resolving and preventing issues. |
Documenting the issue and often leading to a CAPA if deemed necessary. |
Cross-Contamination vs. Mix-Up
Cross-Contamination
The transfer of bacteria, other microorganisms, or other substances from one substance or object to another, especially when it's harmful.
Mix-Up
This refers to a mistake in which items or processes are incorrectly matched, combined, or confused with one another.
|
Criteria |
CAPA | Deviation |
|
Definition |
The transfer of harmful substances or microorganisms from one to another. |
A mistake where items or processes are incorrectly matched or combined. |
|
Concern |
Primarily associated with the transfer of harmful agents. |
Mainly relates to the incorrect matching or combination of items. |
|
Identification |
Often detected through quality tests, inspections, or when product defects or adverse effects arise. |
Often identified during inspections, audits, or product reviews. |
|
Prevention |
Sterilization, sanitation, controlled environments, and proper procedures. |
Proper labeling, SOPs, training, and organizational measures. |
Master Batch Record vs. Batch Production Record
Master Batch Record
A document that provides the detailed steps and procedures for producing a specific product batch size. It acts as a template from which the batch production record is generated.
Batch Production Record (BPR)
A record of a single batch of a product that is produced, detailing the actual ingredients used, processes followed, and controls applied. It is based on the Master Batch Record for that product.
|
Criteria |
Master Batch Record | Batch Production Record |
|
Definition |
A template providing detailed steps and procedures for producing a specific product batch size. |
A record detailing the actual production and control of a single batch. |
|
Purpose |
To ensure consistency in production and standardization across batches. |
To provide a detailed account of each specific batch produced. |
|
Content |
Provides theoretical quantities, processes, and instructions. |
Contains actual quantities used, actual results, deviations, and signatures. |
|
Usage |
Used as a blueprint for producing all batches of a product. |
Used to document the production of a specific batch. |
|
Modification |
Changes are typically subject to regulatory review or approval and are controlled by the Quality department. |
Can have annotations or deviations specific to the batch, but is based on the unchanging MBR. |
Shelf Life vs. Expiration Date
Shelf Life
Is the length of time for which an item remains usable, suitable for consumption, or saleable. It indicates how long a product can be expected to retain its intended quality under specified conditions.
Expiration Date
The specific date after which a product is no longer guaranteed to be safe or effective and should not be used.
|
Criteria |
Master Batch Record | Batch Production Record |
|
Definition |
Duration a product remains usable or maintains its intended quality. |
Specific date after which the product should not be used. |
|
Purpose |
To provide a timeframe for optimal product quality. |
To indicate the end of the guaranteed product quality or safety. |
|
Based on |
Stability studies, real-time data, and accelerated aging tests. |
Real-time data and predetermined criteria. |
|
Format |
Typically given as a length of time (e.g., 24 months). |
Usually provided as a specific day, month, and year. |
|
Implication |
The product is expected to retain its quality until the end of its shelf life under stated conditions. |
Products may no longer be safe or effective after this date. |
Industry-Specific Quality Concepts: Medical Device
Biocompatibility vs. Sterilization Validation
Biocompatibility
The ability of a material to perform with an appropriate host response in a specific application. It ensures that when medical devices come in contact with the body, they do not produce an immune rejection or any adverse local or systemic effects.
Sterilization Validation
A documented procedure is used to confirm that the sterilization process, whether it is physical or chemical, can consistently deliver a product that meets all specified requirements. It ensures the effective elimination or deactivation of all viable organisms.
|
Criteria |
Biocompatibility | Sterilization Validation |
|
Definition |
Compatibility with living tissues. |
Ensures the effectiveness of a sterilization process. |
|
Purpose |
Ensures device safety in contact with the body. |
Ensures consistent product sterility. |
|
Testing |
Involves biological evaluations and chemical assessments. |
Involves microbial, physical, and chemical process assessments. |
Critical Quality Attribute (CQA) vs. Critical Quality Parameter (CQP)
CQA (Critical Quality Attribute)
A physical, chemical, biological, or microbiological property or characteristic that should be within an approved limit, range, or distribution to ensure the desired product quality.
CQP (Critical Quality Parameter)
Parameters relating to raw materials, production processes, or the product itself, that have a direct impact on the quality attributes of the finished product.
|
Criteria |
CQA | CQP |
|
Definition |
Physical or chemical properties to ensure product quality. |
Parameters impacting finished product quality. |
|
Application |
Various manufacturing processes for quality assurance. |
Raw materials, production processes, and the product itself. |
|
Objective |
Ensure the product remains within specified quality limits. |
Ensure parameters are controlled for quality outcomes. |
Design Input vs. Design Output
Design Input
The physical and functional requirements of a product are based on user needs, regulatory requirements, and other sources.
Design Output
The results of the design and development phase in terms of specifications, drawings, process parameters, and other necessary documentation.
|
Criteria |
Design Input | Design Output |
|
Definition |
Physical and functional product requirements. |
Results and documentation of the design process. |
|
Based on |
The user needs regulatory requirements. |
Design inputs and design processes. |
|
Outcome |
What the product should achieve or feature. |
How the product meets or achieves the design inputs. |
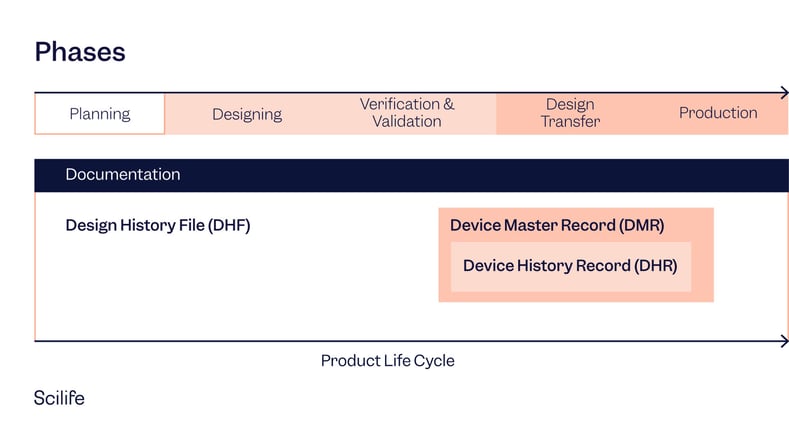
Device Master File (DMR) vs. Device History Record (DHR) vs. Design History File (DHF)
DMR (Device Master File)
A compilation of records that contains the procedures and specifications for a finished device.
DHR (Device History Record)
A compilation of records that contains the production history of a finished medical device.
DHF (Design History File)
A compilation of records that describes the design history of a finished medical device.
|
Criteria |
DMR | DHR | DHF |
|
Definition |
Procedures and specs for a device. |
Production history of device batches. |
Design history of a device. |
|
Purpose |
Ensure the device is manufactured to design specifications. |
Trace and ensure compliance with device production. |
Document and ensure design process compliance. |
|
Content |
Specifications, drawings, and process parameters. |
Dates, quantities, and device labels for each production run. |
Design plans, reviews, verifications, and validations. |
Harm vs. Hazard
Harm
The physical injury or damage to the health of people, or damage to property or the environment. (“ISO/IEC Guide 63:2019”)
Hazard
The potential source of harm. (“ISO/IEC Guide 63:2019”).
|
Criteria |
Harm | Hazard |
|
Definition |
Physical injury or health damage. |
The potential source of harm. |
|
Nature |
Consequence or outcome. |
Source or situation. |
|
Example |
Cut from a sharp object. |
The sharp object itself. |
ISO 9001 vs. ISO 13485 vs. FDA 21 CFR Part 820
ISO 9001
A quality management system standard that sets out the criteria based on various quality management principles including strong customer focus and continual improvement. It was developed by the International Organization for Standardization (ISO) and first released in 1987. It is one of several documents in the ISO 9000 family.
ISO 13485
A quality management system standard specifically for the medical devices industry that sets out the criteria based on various quality management principles emphasizing risk management and regulatory requirements. It was developed by the International Organization for Standardization (ISO).
FDA 21 CFR Part 820
US regulations for quality system regulations about the methods, facilities, and controls used for the design, manufacture, packaging, labeling, storage, installation, and servicing of all finished medical devices.
|
Criteria |
ISO 9001 | ISO 13485 | FDA 21 CFR Part 820 |
|
Focus |
Quality management in general. |
Quality management for medical devices. |
Regulations for the quality system of medical devices in the US. |
|
Emphasis |
Customer focus, continual improvement. |
Risk management, regulatory requirements. |
Manufacturing, design, and servicing of medical devices. |
|
Usage |
Applicable to any industry. |
Specifically for the medical device industry. |
US-based medical device manufacturers. |
PMS vs. PMCF vs. PSUR
PMS (Post-Market Surveillance)
A systematic process that is used by medical device manufacturers to proactively collect and analyze experiences from the marketed medical devices to identify any adverse events or trends.
PMCF (Post-Market Clinical Follow-Up)
Refers to a continuous process that updates the clinical evaluation and is used to ensure the continued safety and performance of the medical device throughout its expected lifetime.
PSUR (Periodic Safety Update Reports)
Refers to regularly scheduled reports that provide an update on the safety of a pharmaceutical drug or a medical device based on data collected from various sources, including clinical trials and post-marketing studies.
|
Criteria |
PMS | PMCF | PSUR |
|
Purpose |
Monitor marketed medical devices. |
Update clinical evaluation of medical devices. |
Update on drug/device safety. |
|
Focus |
On adverse events, and trends. |
Continued safety and device performance. |
Data from clinical trials and post-marketing studies. |
|
Frequency |
Ongoing process. |
Continuous process. |
Regularly scheduled intervals. |
Product Specification vs. Process Specification
Specification
A document that states the requirements to which a given product or service must conform.
Product Specification
A detailed description of the requirements, characteristics, and expected performance of a specific product.
Process Specification
A detailed description of the procedures, inputs, outputs, and controls required to consistently produce a specific product.
|
Criteria |
Product Specification | Process Specification |
|
Definition |
Details of a product's requirements. |
Details of procedures to produce a product. |
|
Focus |
End product. |
Procedures and methods. |
|
Example |
Dimensions, materials, performance. |
Steps, temperatures, tools, and duration. |
Process validation vs. Design validation
Validation
The act of confirming a product or service that meets the requirements which was intended.
Process validation
Establishing objective evidence that a process consistently produces a result or product meeting its predetermined specifications and quality attributes.
Design validation
Establishing objective evidence that device specifications conform to user needs, intended uses, and any specific regulatory requirements.
|
Criteria |
Process Validation | Design Validation |
|
Definition |
Confirms process consistency. |
Confirms design meets user needs. |
|
Focus |
Production process. |
Design output. |
|
Stage |
After product design. |
After design and before commercial production. |
Risk-Benefit Analysis vs. Risk Analysis
Risk-Benefit Analysis
Refers to the evaluation of the positive outcomes (benefits) against the potential negative outcomes (risks) of taking a particular action or intervention.
Risk analysis
A systematic use of available information to identify hazards and to estimate the risk
(“ISO/IEC Guide 63:2019”).
|
Criteria |
Process Validation | Design Validation |
|
Definition |
Evaluate benefits against risks. |
Estimates the risk from potential hazards. |
|
Focus |
On positive and negative outcomes. |
On potential sources of harm. |
|
Outcome |
Decision about an action or intervention. |
Identification and estimation of risks. |
Supplier Audit vs. Supplier Evaluation
Supplier Audit
Refers to a formal review of a supplier's processes, quality systems, and capabilities to ensure they meet the requirements of the contracting organization.
Supplier Evaluation
An assessment of a supplier's ability to deliver goods or services in terms of quality, timeliness, cost, and other relevant factors.
|
Criteria |
Supplier Audit | Supplier Evaluation |
|
Definition |
Formal review of supplier's processes. |
Assessment of supplier's delivery ability. |
|
Focus |
On compliance and capability. |
On quality, timeliness, cost, etc. |
|
Outcome |
Report with findings and recommendations. |
Decision on supplier selection or continuation. |
Unique Device Identifier (UDI) vs. Universal product code (UPC)
Unique device identifier (UDI)
A series of numeric or alphanumeric characters that is created through a globally accepted device identification and coding standard, unique to a specific medical device. It adequately identifies a device through its distribution. UDI is composed of:
- A device identifier – a mandatory, fixed portion of a UDI that identifies the specific version or model of a device and the label of that device.
- A production identifier – a conditional, variable portion of a UDI that identifies one or more of the following when included on the label of the device
Universal product code (UPC)
A 12-digit barcode symbology that is used in the retail industry to identify a specific product and its manufacturer.
|
Criteria |
UDI | UPC |
|
Application |
Medical Devices |
Retail products |
|
Focus |
On global identification of medical devices. |
On identification of retail products and manufacturers. |
Sum up
Now you have an idea about the most common quality concepts that every life science industry knows and uses.
You can always surf on the Scilife Library and Scilife Glossary pages to get deeper information about specific concepts.
Note
The definitions and differences are based on general industry understanding as some of the terms were not explicitly defined or have slight nuances or differences in the corresponding regulations.





