

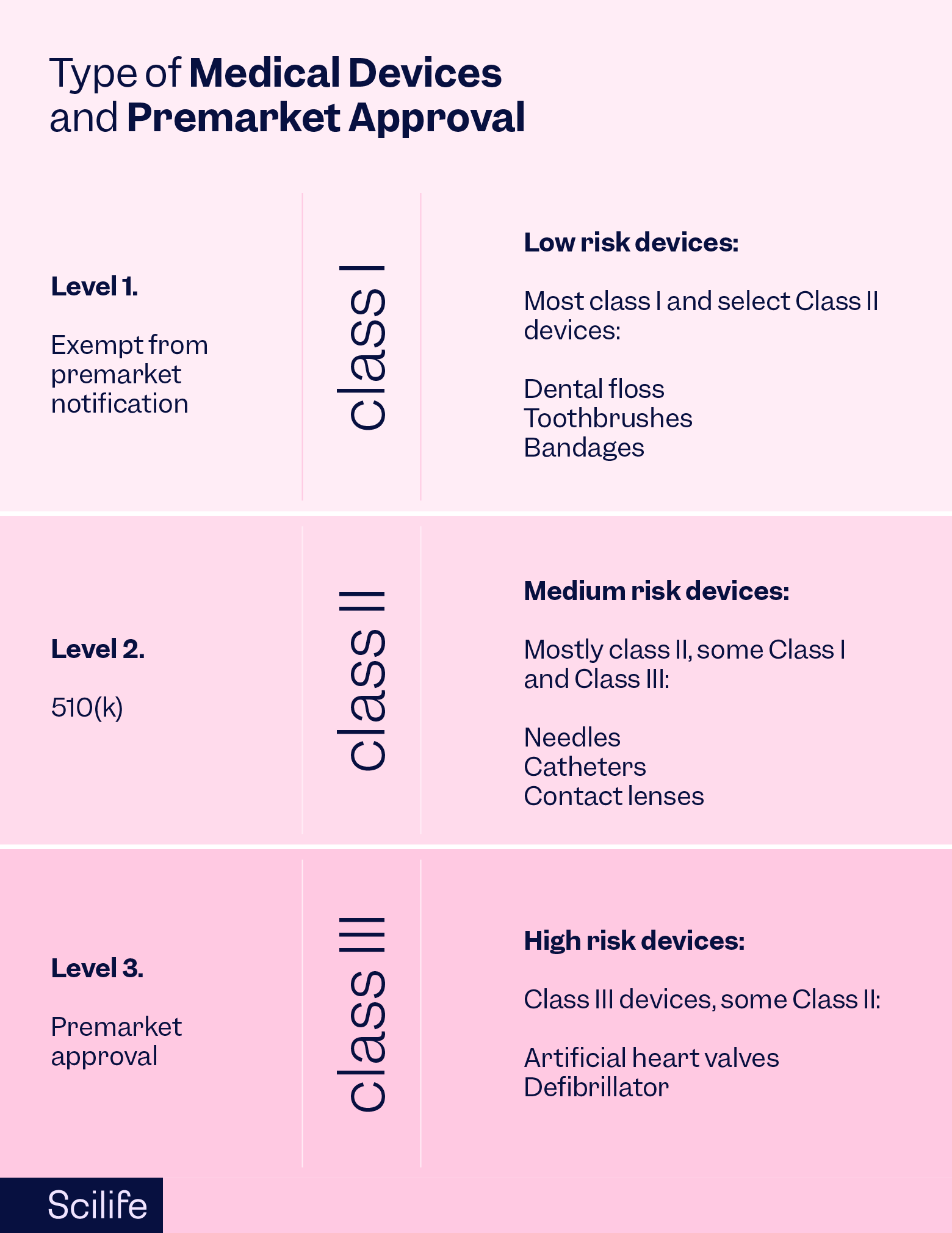
The United States Food and Drug Administration (FDA) is the regulatory body responsible for the safety and efficacy of medical devices in the United States. The 510(k) clearance is their classic premarket clearance for Class II medical devices that most of us are probably familiar with. The 510(k) demonstrates that devices are substantially equivalent to similar devices on the market, clearing them for sale in the US.
Class III devices, on the other hand, are subject to the most stringent type of device marketing application required by the FDA, the Premarket Approval (PMA).
In this article, we'll review the basics of the PMA, who can submit a PMA application, what information is required, and how the PMA process works.
What type of Medical Devices need a PMA?
PMAs are typically required for Class III medical devices, considered high-risk devices that support or sustain human life, are of substantial importance in preventing impairment to human health, or present a potential, unreasonable risk of illness or injury. The PMA is the most stringent type of market approval scheme in the US due to the high risk associated with Class III medical devices. Any malfunctions or adverse events with these devices can lead to significant harm to the patient.
Some examples of Class III medical devices include implantable cardiac pacemakers, artificial heart valves, blood vessel stents, and certain implantable neurostimulators.
Who can submit a PMA?
Medical device manufacturers or sponsors can submit PMA applications. During the PMA process, manufacturers are responsible for ensuring that their devices meet the FDA's rigorous standards for safety and effectiveness. Additionally, manufacturers should conduct clinical trials to gather clinical data supporting their PMA submission.
It's important to note that the PMA process can be time-consuming and costly, often requiring substantial financial investment and a dedicated team of experts to navigate the regulatory requirements. Therefore, typically, only larger medical device companies or manufacturers have the resources to undertake the PMA process.
Types of PMAs
There are four types of PMAs, each with different applications and advantages and disadvantages.
1. Traditional PMA
The traditional PMA is, ahem, the traditional method of submitting your PMA to the FDA. It consists of many volumes of material submitted to the FDA, much like the European technical file. The entire PMA is submitted at once because all clinical testing has already been completed, and the device has already been approved in markets with established medical device regulations, such as the EU. It is not the quickest way to get a device to market, but if all the documentation and clinical investigations are already complete, it makes sense for the manufacturer.
2. Modular PMA
The modular PMA deconstructs the complete PMA submission into modules, such as preclinical, clinical, and manufacturing, which are submitted as soon as the manufacturer completes each module. Over time, the PMA is completed, module by module. The FDA reviews each module as completed, allowing for a quicker PMA closure once the last module has been submitted.
The modular PMA is recommended for devices in the early stages of clinical studies, not for devices that are close to clinical investigation completion or if the device design is likely to change in the future.
3. Product development protocol
For the product development protocol PMA submission, the manufacturer comes to an early agreement with the FDA about the activities that should be performed to demonstrate the safety and effectiveness of a device. Discussing the upcoming regulatory activities with the authority allows device manufacturers in the development cycle to collaborate with the FDA on the testing to be performed, thus preempting wasted resources on activities that might not please the FDA. The product development protocol is a contract between the manufacturer and the FDA, establishing milestones, development activities, outputs, and acceptance criteria for their device development. The manufacturer can always move forward with their development protocol in their own time as long as the FDA is informed.
The product development protocol is recommended for medical devices with well-established technologies in device development.
4. Humanitarian device exemption
Humanitarian Use Devices are medical devices intended to treat or diagnose diseases that impact less than 8,000 people annually in the United States. Humanitarian Use Devices are exempt from the regular PMA and are subject to certain profit and use restrictions.
Required Data and Information
The PMA application is a comprehensive document that includes a vast amount of data and information to support the medical device's safety and effectiveness.
Some of the essential components of a PMA application include:
- Device Description: A detailed description of the medical device, including its design, materials, and intended use.
- Preclinical Data: Information on laboratory and animal testing to evaluate the device's safety.
- Clinical Data: Results of clinical trials demonstrating the device's safety and effectiveness in humans.
- Manufacturing Information: Detailed information about the device's manufacturing processes, quality control, and quality assurance.
- Risk Analysis: A comprehensive analysis of the device's potential risks and the steps to mitigate them.
- Labeling and Instructions for Use: Clear and informative labeling and instructions for healthcare professionals and patients.
- Proposed Indications for Use: A description of the specific conditions or diseases the device is intended to treat or diagnose.
The PMA Application Process Explained
The PMA process can be divided into several key stages:
- Preparation: Manufacturers gather the necessary data and information, conduct clinical trials, and ensure their device complies with FDA regulations.
- Submission: The manufacturer submits the digital PMA application to the FDA's Center for Devices and Radiological Health (CDRH).
- Administrative and limited scientific review: The FDA conducts an acceptance review to ensure the application is complete and meets the regulatory requirements.
- In-depth review: FDA experts assess the submitted data, including preclinical and clinical results, to determine the device's safety and effectiveness.
- Inspection: FDA inspectors may conduct on-site visits to the manufacturer's facilities to verify the information in the application.
- Panel review: In some cases, an FDA advisory panel may be convened to provide expert advice on the device.
- Final Decision: Based on the review, the FDA decides to approve or deny the PMA application.
Do you want to dive deeper into the Regulatory Framework of Medical Devices in the US? Check our training below.
Conclusion
The Pre-Market Approval process is the most critical step in ensuring the safety and efficacy of high-risk medical devices in the United States. It requires extensive data and rigorous evaluations to ensure these devices are safe for patients and meet the highest standards.
While the PMA process can be daunting, it's essential for the health and well-being of patients, not to mention the regulatory approval required for market authorization in the US. PMAs typically take several years to complete and receive FDA approval. Once the PMA application is approved, the device can be marketed for its intended use.





