

Understanding current Good Manufacturing Practices (cGMP) in the pharmaceutical industry can be challenging. That’s why quality professionals in the pharma industry actively seek to understand cGMP compliance. In most cases, they also learn on the go as they start implementing those requirements.
In this deep dive, we explain how cGMP forms the foundation of pharmaceutical product quality. If you want to learn how cGMP connects to other key pharmaceutical regulations, take a look at our QMS in pharma guide.
Key takeaways
What are cGMP compliance requirements? cGMP FDA guidance
The FDA’s current Good Manufacturing Practices (cGMP) regulate manufacturing processes, facility design, monitoring, controls, and cGMP compliance to help ensure the identity, strength, quality, purity, and safety of drug products.
Establishing rigorous operating procedures, acquiring acceptable quality raw materials, investigating product quality variations, and maintaining trustworthy testing labs are all part of this.
If properly implemented, this formal system of controls at a pharmaceutical company aids in preventing instances of contamination, confusion, divergence, failure, and error. This helps ensure that drug products consistently meet established quality standards.
The cGMP FDA guidance rules were created with flexibility in mind, allowing each producer to choose how to best implement the required controls by utilizing design, processing, and testing techniques that are based on good scientific principles. Due to the regulations’ flexibility, businesses can employ cutting-edge methods and technology to continuously improve quality.
What is the “c” in cGMP?
The “c” in cGMP is often also considered to stand for “contemporary,” because it reflects the expectation that manufacturers use current, appropriate systems, processes, and technologies to abide by the rules.
To avoid contamination, confusion, and errors, systems and equipment that may have been top-of-the-line 10 or 20 years ago may now be deemed less than acceptable. It is crucial to remember that the cGMPs are only the necessary minimums. Many pharmaceutical factories have already adopted thorough, contemporary quality systems and risk management strategies that go above and beyond these minimum requirements.
The FDA’s portion of the CFR (Code of Federal Regulations) can be found in Title 21. The pharmaceutical or drug quality regulations are addressed in several parts of Title 21, including parts 1-99, 200-299, 300-499, 600-799, and 800-1299.

These are the 21 CFR parts that explain the cGMP requirements:
-
21 CFR Part 314 - FDA approval to market a new drug
-
21 CFR Part 210 - cGMP in Manufacturing Processing, Packing, and Holding of Drugs
-
21 CFR Part 211 - cGMP for Finished Pharmaceuticals
-
21 CFR Part 212 - cGMP for Positron Emission Tomography Drugs
-
21 CFR Part 600 - Biological Products
Additionally, the following pages provide cGMP requirements for particular product types and manufacturing considerations:
-
Field Alert Reports (for NDAs and ANDAs)
-
Biological Product Deviation Reports (for BLAs)
While this article focuses on FDA cGMP guidance (21 CFR Parts 210 and 211), pharmaceutical manufacturers operating globally must also comply with other regulatory frameworks.
In the European Union, GMP requirements are defined in EudraLex Volume 4, while international guidelines from the International Council for Harmonisation (ICH), such as ICH Q7 (APIs), Q8 (Pharmaceutical Development), Q9 (quality risk management), and Q10 (pharmaceutical quality system), help harmonize expectations across regions.
The origins of GMP regulations in the GxP: Scilife Academy course


Why is cGMP so important in the pharmaceutical industry?
Pharmaceutical products have the highest risks to human health. This is why drug manufacturing facilities need to be regulated by the latest cGMP and other relevant regulations to assure the end-users that a drug product is safe and will work.
Even though cGMPs require manufacturers to test each batch, it is not possible to ensure the expected quality by only testing the batch.
The more critical part is manufacturing those drug products under the conditions and practices required by the latest cGMP regulations to guarantee that quality is built into the design at every step of the manufacturing process. In this way, your company provides that:
-
Manufacturing facilities in pharma are in good condition and designed, monitored, and controlled properly.
-
Drug products meet the identity, strength, purity, and quality specifications.
-
Equipment is properly maintained and calibrated.
-
Employees who are qualified and fully trained.
-
Processes that are reliable and reproducible.
Those are just a few examples of how cGMP requirements help assure drug product safety and efficacy.
What is the use of cGMP in pharma?
Consumers have no means of knowing whether the substance they are ingesting is safe and effective, and testing alone cannot guarantee quality. This is where cGMP in pharma comes into play: medications must be manufactured in compliance with the conditions and procedures required by the cGMP rules to guarantee that quality is incorporated into the design and manufacturing process at every level.
Maintaining facilities, equipment that are correctly calibrated, personnel who are qualified and trained, and processes that are dependable and repeatable are just a few examples of such conditions and practices.
The cGMP guidelines include the following aspects to take into account:
-
Maintain clean and sanitary manufacturing facilities.
-
Ensure controlled environments to prevent cross-contamination that could compromise product quality.
-
Define and strictly control manufacturing procedures, with validation of critical processes to ensure consistency and compliance.
-
Approve and document any changes that may impact product quality or efficacy in accordance with established procedures.
-
Provide regular training to personnel on procedures and ensure accurate, real-time documentation of manufacturing activities (whether manual or digital).
-
Investigate and document all deviations.
-
Maintain complete production and distribution records to ensure full batch traceability.
-
Protect product quality during storage and distribution.
-
Establish and maintain an effective recall system to enable prompt removal of any batch from the market.
-
Investigate quality defects and complaints, and implement corrective and preventive actions (CAPA) to prevent recurrence.
Staying cGMP-compliant gets much easier when documents, training, workflows, and quality events work together. A GMP-compliant eQMS like Scilife makes that possible.
Contamination control and Annex 1 updates
Preventing contamination has always been a core principle of cGMP, but recent regulatory updates have significantly strengthened expectations. The revised EU GMP Annex 1 (effective from 2023–2025) introduces stricter requirements for sterile manufacturing and places strong emphasis on the implementation of a Contamination Control Strategy (CCS).
A CCS is a holistic, risk-based approach that identifies and controls potential contamination risks across facilities, equipment, personnel, processes, and utilities.
While Annex 1 is a European regulation, its principles are increasingly influencing global regulatory expectations and inspection practices.
cGMP regulations and definitions
cGMP regulations for finished pharmaceutical products are defined under FDA 21 CFR Part 211 – cGMP for Finished Pharmaceuticals. These standards consist of the following subparts:
Subpart A - General provisions
Minimum requirements of current good manufacturing practices for the preparation of drug products for human and veterinary use.
Subpart B - Organization and personnel
Requirements for ensuring that employees are fully trained and qualified and that their responsibilities are explicitly defined.
Subpart C - Buildings and facilities
Requirements for ensuring that buildings and facilities are in good condition in terms of:
-
Design and construction features
-
Lighting
-
Ventilation, air filtration, heating, and cooling
-
Plumbing
-
Sewage (liquid waste) and refuse (solid waste)
-
Washing and toilet facilities
-
Sanitation
-
Maintenance
Subpart D - Equipment
Requirements for ensuring that equipment is adequately constructed, cleaned, maintained, and calibrated.
Subpart E - Control of components, containers, and closures of drug products
Requirements for describing sufficient details of receipt, identification, storage, handling, sampling, testing, approval, rejection of components and containers, and closures.
Handling and storage requirements for components, drug product containers, and closures to prevent contamination.
Identification and tracking of each container or a group of containers for components or containers or closures with a unique code for each batch in each shipment received.
Subpart F - Production and process controls
Requirements for ensuring that production and process control are designed to guarantee that the drug products have identity, strength, quality, and purity. In addition, processes and procedures should be reproducible and reliable.
Subpart G - Packaging and labeling control
Requirements for correct labels, labeling, and packaging materials for drug products.
Subpart I - Laboratory controls
Requirements to ensure laboratory controls, including establishing scientifically sound and proper specifications, standards, sampling plans, and testing procedures.
Details on components, containers, closures, in-process materials, labeling, and drug products conform to standards of identity, strength, quality, and purity.
Subpart J - Records and reports
This section outlines record retention requirements for components, containers, closures, labeling, and production, control, and distribution activities.
It also states that these records must be maintained on-site and be easily accessible for authorized inspections during the entire retention period.
Subpart K - Returned or salvaged drug products
Requirements for identifying and holding returned drug products and those subjected to improper storage conditions.

Data integrity and ALCOA+ principles
Data integrity is a fundamental pillar of cGMP compliance and a major focus of regulatory inspections worldwide. Pharmaceutical companies must ensure that all data generated throughout the product lifecycle is complete, consistent, and reliable.
Regulators commonly refer to the ALCOA+ principles to define good data practices.
Data should be:
-
Attributable (linked to the person who generated it)
-
Legible (readable and permanent)
-
Contemporaneous (recorded at the time of the activity)
-
Original (or a verified true copy)
-
Accurate
The “+” expands this to include that data should also be complete, consistent, enduring, and available.
Ensuring data integrity requires robust processes, controlled systems, audit trails, and proper training, especially when using digital tools such as eQMS platforms.
Common cGMP violations under cGMP FDA guidance
A company may recall a product if the failure to comply with cGMP results in the distribution of a drug that, for example, contains too little active ingredient and does not provide the benefit claimed on the label. Taking these medications off the market safeguards the general public from additional harm.
Although the FDA cannot compel a manufacturer to recall a medicine, most do so voluntarily or at the FDA’s request. The FDA has the authority to issue a public alert and can confiscate a medicine if a manufacturer refuses to recall it.
Even in cases when there is no clear evidence that an error is impairing a drug’s effectiveness, the FDA is nonetheless permitted to file a seizure or injunction case in court to resolve cGMP violations. When the FDA does file a seizure action, the agency requests a court order allowing federal agents to seize the adulterated pharmaceuticals. At this time, the FDA also requests that the court order the corporation to cease violating cGMP.
Court orders requiring businesses to take numerous actions to address cGMP violations are frequently issued in both seizure and injunction cases. These actions might include fixing facilities and equipment, enhancing sanitation and cleanliness, carrying out additional tests to confirm quality, and improving employee training.
Due to cGMP violations, the FDA may even file criminal charges and impose fines.
As regulatory expectations evolve, particularly around data integrity, traceability, and risk management, many companies rely on digital systems to support and sustain cGMP compliance.
FDA warning letters in the pharmaceutical industry - the top trends and violations.

What are the key concepts of quality systems under cGMP compliance?
The following key concepts are critical for modern quality systems in the pharmaceutical industry.
Quality
Quality characteristics such as identity, strength, and purity should be established in a pharmaceutical product to ensure the mandated level of safety and effectiveness.
Quality by Design and product development
The strategy, Quality by Design, is used when designing and developing a product and associated manufacturing processes to ensure that the finished product consistently meets the defined quality at the end of the manufacturing process.
Quality Risk Management
Quality risk management can support manufacturers in establishing process parameters and specifications required to produce a drug, then identify and mitigate risks of any change in the process or specifications.
CAPA (Corrective and Preventive Action)
Corrective and preventive actions focus on investigation, correction of discrepancies, and prevention of recurrence.
Change control
Change control focuses on managing change to prevent unintended consequences. Major manufacturing changes, such as altering specifications, a critical product attribute, or bioavailability, require regulatory documentation before regulatory approval.
The quality unit
Most modern quality system concepts have a correlation with the cGMP regulations. Current industry practices generally divide the responsibilities of the Quality Control Unit (QCU) between Quality Control (QC) and Quality Assurance (QA) functions.
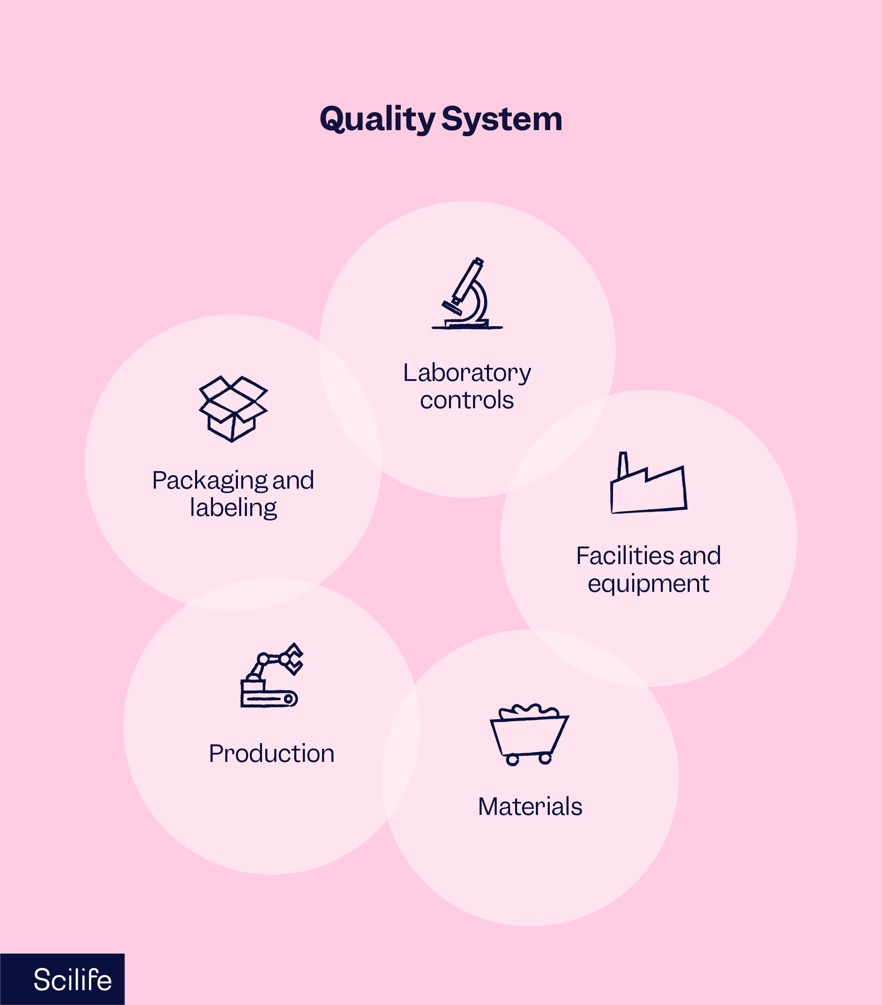
Six-system inspection model
The FDA's Drug Manufacturing Inspection Compliance Program contains instructions to FDA personnel for conducting inspections.
It is a systems-based approach to inspection and is consistent with a robust quality system model.
The diagram shows the relationship between the six systems: the quality system plus the five manufacturing systems. The quality system provides the foundation for the linked manufacturing systems and functions.
What is a GMP certificate?
Each manufacturer must stay compliant with its processes and implement the systems according to the latest GMP.
GMP certification and proof of compliance vary depending on the regulatory authority. In the European Union, national competent authorities may issue a GMP certificate following a successful inspection of a manufacturing site.
In the United States, however, the FDA does not issue GMP certificates. Instead, compliance with cGMP is assessed through inspections, with outcomes documented in inspection reports and classification statuses (e.g., NAI, VAI, OAI).
For all manufacturers, maintaining GMP compliance is an ongoing responsibility. Regulatory authorities may re-inspect facilities periodically, especially after significant changes to processes, equipment, or facilities.
Rather than a one-time certification, cGMP compliance should be viewed as a continuous commitment to quality, risk management, and improvement.
How to get GMP certified step by step in the US and the EU.
How can software help us comply with and manage cGMP requirements?
Let’s take a closer look at how software can help companies manage some core cGMP requirements more efficiently.
Organization and personnel
Training key personnel to perform their responsibilities in a pharmaceutical company is required by cGMP in pharma. In addition, pharma companies should retrain staff on any changed procedures.
If your company uses QMS software (eQMS) with a training feature like Scilife, all relevant personnel automatically receive training tasks based on their job roles, along with a training plan for each job role. In addition, employees will automatically be notified when a training task is assigned.
Facilities and equipment
The building and facilities of a pharmaceutical company should be appropriately established and maintained to guarantee all manufacturing conditions remain safe and effective. Equipment should stay well-maintained and calibrated.
An equipment calibration plan within an eQMS can allow you to track and manage all equipment calibrations. The software can automatically notify responsible personnel ahead of upcoming calibration activities and assign tasks to the relevant employees.
Production process and procedures
If you still use older technology and dated software, your company may struggle to meet current cGMP expectations. Make sure to review all procedures regularly and verify that you use the latest science and technology for your products.
For example, setting up an automated annual review reminder for your forms will allow relevant employees to receive notifications before the next review.
Conclusion
cGMP is ultimately about protecting patients. By enforcing these standards, regulators like the FDA help ensure that pharmaceutical products are safe, effective, and consistently manufactured.
Real-world failures show what’s at stake. For instance, the 2012 fungal meningitis outbreak linked to the New England Compounding Center is a stark reminder that gaps in quality systems can have serious (and sometimes fatal!) consequences. Properly implemented cGMP should therefore not be seen as a regulatory burden, but as a critical safeguard.
At its core, cGMP compliance relies on a few essential elements: well-maintained and calibrated equipment, qualified and trained personnel, and robust, repeatable processes.
But strong internal processes alone are not enough. Pharmaceutical quality also depends on the reliability of suppliers. Without proper supplier oversight and quality agreements, risks can enter the system before manufacturing even begins. That’s why having visibility into your suppliers’ controls and building strong, collaborative relationships with them is key to maintaining a resilient quality system.





