

In the European Union, life sciences companies that export to the United States must comply with 21 CFR Part 11, the FDA regulation that oversees electronic records and electronic signatures. As digital systems continue to replace traditional paper-based processes, understanding this regulation has never been more important.
In this article, we’ll take a clear and practical deep dive into 21 CFR Part 11 compliance and digital signatures. You’ll learn what the regulation covers, when it applies, which systems it affects, and how electronic signatures must be handled.
If you’re also interested in broader pharmaceutical compliance topics, we recommend checking out our QMS in pharma guide as a complement to this overview.
Introduction to 21 CFR Part 11
Prior to diving deeper into 21 CFR Part 11 compliance and digital signatures, we discussed the meaning of electronic records and audit trails in previous articles. We recommend that you take a look at these articles to build your foundations!
Both electronic records and audit trails involve the use of electronic signatures, which are key to improving productivity in the regulated quality control laboratory. This improved productivity can help companies in life sciences release products to market faster.
And whether your organization uses digital tools or is still transitioning to electronic systems, understanding both the EU GMP Annex 11 and 21 CFR Part 11 guidelines is essential.
In this article, we will focus specifically on compliance with the 21 CFR Part 11 electronic records and electronic signature requirements, but you can dive into our recommended learning below to learn about EU GMP Annex 11!
Your guide to electronic records and signatures according to EU GMP Annex 11.

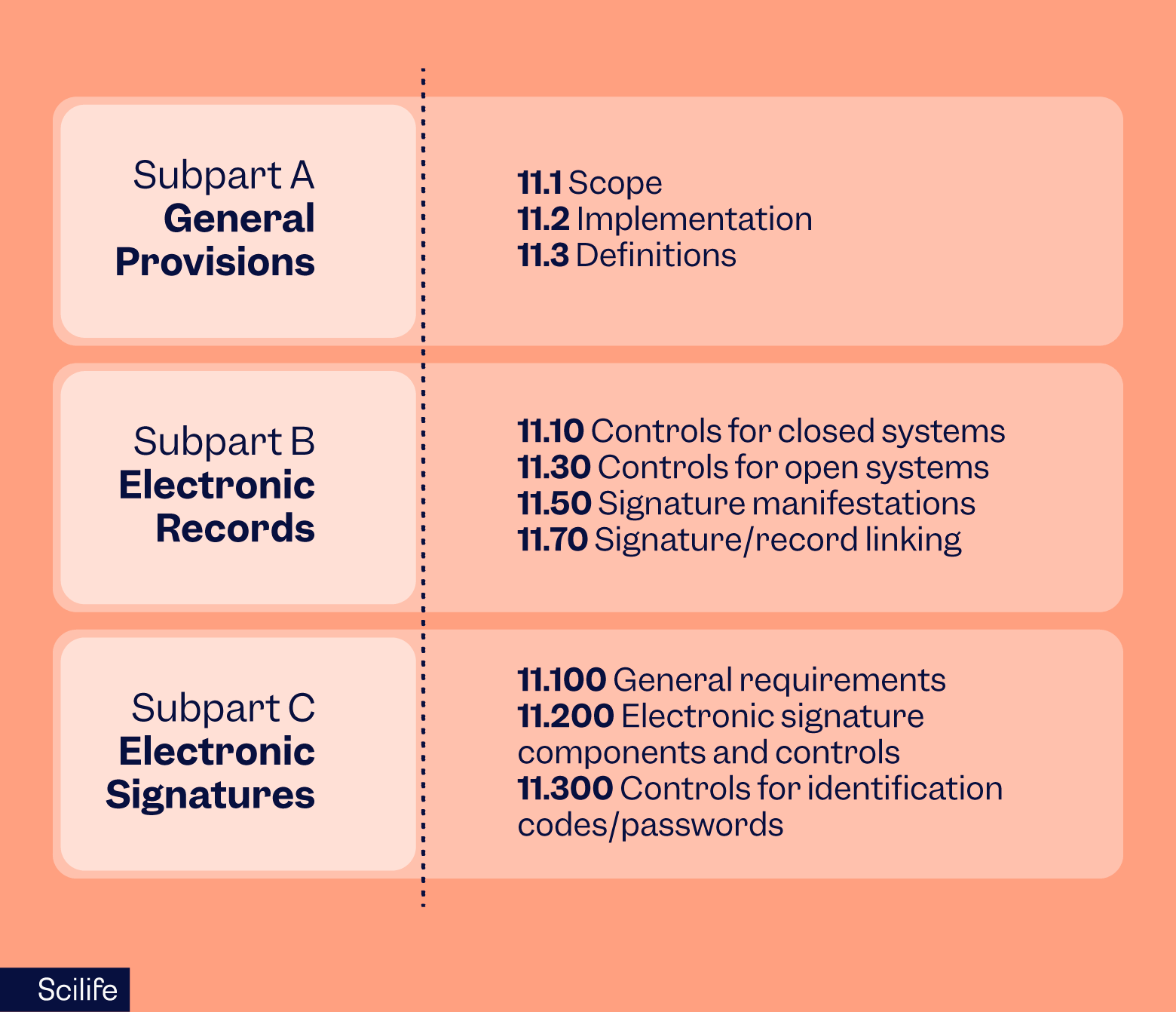
What is 21 CFR Part 11?
21 CFR Part 11 is part of Title 21 of the Code of Federal Regulations (CFR), which is the section that governs food and drugs in the United States.
This part defines the requirements for electronic records and electronic signatures used by pharmaceutical, biotechnology, and medical device manufacturers when submitting information to the U.S. FDA. It establishes the criteria under which these electronic records and signatures are accepted, ensuring they are as trustworthy, reliable, and legally binding as their paper-based counterparts.
It’s important not to confuse Part 11 with Part 211, which relates to current Good Manufacturing Practices (cGMP) for finished pharmaceuticals. While Part 211 focuses on manufacturing quality, Part 11 addresses the integrity and authenticity of electronic records and signatures.
To better understand the regulation’s name:
- 21 stands for “Title 21.” This section of the overarching CFR regulation applies to food and drugs.
- CFR is short for the “Code of Federal Regulations.” This is a coded set of laws published by the United States federal government.
- Part 11 is the scope of the regulation specific to electronic records and electronic signatures, and to the electronic systems used to create, modify, maintain, archive, retrieve, or transmit them.
In practice, Part 11 applies to electronic systems used to create, modify, maintain, archive, retrieve, or transmit regulated records. It ensures that all such records remain authentic, accurate, and tamper-proof throughout their lifecycle.
According to the 21 CFR Part 11, the definitions of electronic record and electronic signature are as follows:
An electronic record includes text, graphics, data, audio, pictorial, and other digital information representations created, modified, maintained, archived, retrieved, or distributed by computers.
An electronic signature consists of a collection of symbols or a series of symbols executed, adopted, or authorized by the individual to serve as legally binding equivalents to their handwritten signatures.
Get a deeper understanding of the topic by checking our training video below:
A brief history of 21 CFR Part 11
The FDA issued the final Part 11 regulation in March 1997, and it became effective on August 20 of that year. However, after implementation, many companies found the requirements overly rigid and difficult to grasp. To address these concerns, the FDA released a guidance document in September 2003 that clarified a more practical, risk-based approach to compliance. This approach to 21 CFR Part 11 compliance and digital signatures emphasized:
- Validation of electronic systems: ensuring systems are fit for their intended use.
- Implementation of electronic audit trails: maintaining traceability of actions and changes.
- Proper archiving and retention of electronic records: keeping records accessible and secure throughout their lifecycle.
Electronic records, 21 CFR Part 11 compliance and digital signatures


Key requirements of 21 CFR Part 11
The key requirements of 21 CFR Part 11 compliance and digital signatures include:
- System validation: the electronic system must be validated to prove it is secure, reliable, and works as intended.
- User access control: only authorized individuals should have access to the system. This includes using unique user IDs and passwords, and revoking access for terminated employees.
- Audit trails: the system must maintain a secure, tamper-proof log of all changes and actions, including who made the change and when.
- Electronic signatures: signatures must be unique, tied to specific records, and link back to the person who signed them. They cannot be copied or transferred.
- Data integrity: electronic records must be accurate, protected from alteration, and stored in a way that ensures their reliability.
- Record management: requirements for the creation, storage, management, and retrieval of electronic records must be followed.

Part 11 electronic records and electronic signatures
The primary reason for 21 CFR Part 11 compliance requirement is security and protection concerns about managing the distribution, storage, and retrieval of records by biotechnology, drug, and medical equipment manufacturers in the digital age.
Originally, the considerable cost of maintaining paper-based filing systems for these companies was intended to satisfy the regulator. A key objective of the new regulation was to enable these firms to adopt paperless systems.
The Guidance for Industry Part 11, electronic records; electronic signatures — Scope and Application guides manufacturers to meet 21 CFR Part 11 compliance by following these steps:
- Validation
- Audit trail
- Legacy systems
- Copies of records
- Record retention
Because Part 11 places strong emphasis on the reliability of electronic signatures, the FDA specifies that every compliant signature must include three essential elements:
- The signer’s full name,
- The date and time when the signer signed, and
- The purpose of the signature such as review, approval, responsibility, or authorship.
Even if you’ve transformed your signature process into a digital platform, these three key pieces of information are still equally important and need to be recorded.
Bear in mind, while Part 11 defines the official regulatory requirements, the FDA’s 2003 Guidance provides a practical, risk-based approach to implementing them. The guidance highlights areas such as validation, audit trails, legacy systems, record copies, and record retention to help companies apply Part 11 in a realistic way.
These ensure that digital systems are reliable, consistent, and legally defensible.
Electronic records for 21 CFR Part 11 software
Part 11 defines electronic records as:
“Any combination of text, graphics, data, audio, pictorial, or other information representation in digital form that is created, modified, maintained, archived, retrieved, or distributed by a computer system.”
An organization’s electronic records may be a part of a closed system or an open system - next up, we define what both concepts are.
Closed systems
A closed system means that access is controlled internally by the people in the company who are responsible for the content of the electronic records.
Organizations that rely on closed systems generally store electronic records exclusively on their own hardware. The records are then accessible on the organization’s internal network via username and password.
Databases with audit trails are a common example of a closed system. Most systems in analytical laboratories are closed as well. Even Scilife represents a closed system.
According to 21 CFR Part 11, Computer System Validation (CSV) and Document Control are essential to ensuring authenticity, integrity, confidentiality, and irrefutability of the records in a closed system.
Open systems
In an open system, the people in the company who are responsible for the system’s electronic records do not control system access. Rather than requiring a username and a password, digital signatures are needed to verify the identities of those who sign any documents.
Open systems can be compliant, but more controls are needed to protect the records from being read, modified, destroyed, or compromised by unauthorized parties. Security and data integrity are usually more challenging in this way.
For example, consider a vendor offering a license to a record-keeping 21 CFR Part 11 software solution. In this case, the vendor controls access to both the software and the records. They may require certain elements of a compliant system, but the system is not inherently compliant. This means that it’s the user’s responsibility to implement the appropriate controls.
21 CFR Part 11 compliance and digital signatures
Traditionally, quality documents like risk assessments, change requests, and training records were printed on paper and handed to each reviewer. They were then approved via wet-ink signature. Electronic signatures are more efficient. Part 11 defines electronic signatures as:
“A computer data compilation of any symbol or series of symbols executed, adopted, or authorized by an individual to be the legally binding equivalent of the individual’s handwritten signature.”
In simple terms, an electronic signature is a simple, legally binding way to authenticate the identity of the person signing a digital document. It is considered the equivalent of a handwritten signature on a paper document.
The FDA approves of electronic signatures over handwritten ones on paper documents if the organization meets certain requirements:
- First, the electronic signature must be unique to one individual.
- Second, the printed name of the signer, along with the date and time, and the signer’s digital signature, should be listed.
- Use two distinct identification components, like username and password.
- And finally, the intent of the signature (e.g., authorship, review, or approval) must be displayed.
When it comes to validating electronic signatures, these aren’t the only considerations. For example, electronic signatures that are not based on biometrics must employ at least two unique identification components, like a username and password for the first login.
To track further activities, password authentication alone is typically sufficient.
That said, the FDA does not permit the sharing of electronic signatures. If the original signer is absent, however, their supervisor and system administrator can jointly sign electronically on their behalf.
Ultimately, though, duplication is prohibited. According to 21 CFR Part 11, no two people can have the same identification code or password, and sharing electronic signatures is not allowed.
Transaction safeguards and loss management procedures can, in this way, prevent issues from compromising your organization’s electronic signatures.

How can I create a password that meets the 21 CFR Part 11 requirements?
Creating passwords that are secure and with 21 CFR Part 11 is key to upkeep security for your electronic records.
To comply with the regulation, your password needs to be integrated in your company’s security system and internal policies. It needs to contain unique IDs, strong authentication, and password complexity to meet the regulation's intent. Here are a few criteria points that the password should satisfy:
- It should contain a minimum of 8 characters, some of them alphanumeric.
- It should not be the same as the previous six passwords.
- It should not be saved in your browser or anywhere else.
- Users should not write down the password anywhere.
- It should not contain full words.
- The password should change every 90 days at least.
- The text should be hidden when typing the password.
- The person who owns the password should be known, so there will be no doubt about who is accessing and editing documents.

Does my software need validation?
Software validation is the systematic process of verifying that a computerized system meets predefined user requirements and operates consistently as intended. It involves thorough testing and documentation to support 21 CFR Part 11 software compliance with regulatory standards like FDA’s 21 CFR Part 11 or ISO 13485.
Many companies use Good Automated Manufacturing Practices (GAMP) while some manufacturers prefer to focus on Part 11 requirements.
What makes a computer system 21 CFR Part 11 compliant?
A 21 CFR Part 11 compliant system should be able to ensure data integrity, security, and confidentiality of electronic records and signatures, while also providing compliance with the requirements outlined in 21 CFR Part 11. Compliant system capabilities include, but are not limited to:
- Detecting any invalid or altered records.
- Generating accurate and complete copies of records in both human-readable and electronic formats.
- Enabling accurate retrieval of records throughout the retention period.
- Limiting system access to only authorized individuals.
- Implementing secure and time-stamped audit trails.
- Linking signatures to electronic records.
- Issuing unique identification codes and passwords.
- Enabling backup and recovery of data in the event of a system failure.
There is also confusion between 21 CFR Part 11 “compliant” and 21 CFR Part 11 “ready” systems, which are distinct states.
21 CFR Part 11-ready refers to a system with features aligning with 21 CFR Part 11, but may need configuration or validation for full compliance.
On the other hand, a 21 CFR Part 11 compliant system means the system has undergone validation and meets the regulation’s requirements. This makes it suitable for FDA-regulated environments without additional modifications.
So, to determine if your software or computer system requires validation, answer the following questions to find out:
What activities does this software cover?
Answer if the software creates, modifies, maintains, archives, retrieves, or transmits records under any requirement outlined in the regulations.
21 CFR Part 11 software using electronic signatures
Does your system request or require user authentication (for example, a username and password) to perform regulated activities or approve documents? If yes, the software falls under Part 11’s electronic signature provisions, and validation must confirm that these signatures are secure, unique, and linked to their corresponding records.
What documentation is needed after review?
Once you determine that validation is necessary for your 21 CFR Part 11 software, your team should prepare documentation demonstrating that the system meets its intended use and Part 11 requirements. This documentation typically includes:
- User requirements specification (URS)
- Validation plan
- Installation qualification (IQ), operational qualification (OQ), and performance qualification (PQ)
- Validation summary report
These documents collectively prove that the system performs consistently and securely within its defined scope.
Are all systems prevalidated?
This depends on the vendor of the system. For example, a system may already be designed to meet 21 CFR Part 11 requirements. In that case, the vendor can give you a documentation/pre-validated validation package so that you can demonstrate how the system complies and significantly decreases the workload of validation activities.
Any life sciences organization must fulfill the requirements of 21 CFR Part 11 or other related regulations to maintain records or submit designated information electronically. Therefore, if you’re planning to transform your paper-based Quality Management System into an electronic QMS, it is essential to know whether it is capable of 21 CFR Part 11 compliance.
How to choose a 21 CR Part 11 compliant QMS
Best practices to comply with 21 CFR Part 11
Achieving and maintaining 21 CFR Part 11 compliance on digital signatures requires more than technology; it demands a disciplined organizational approach.
Best practices focus on creating clear policies, educating staff, validating systems regularly, and documenting processes through SOPs. By institutionalizing these practices, pharmaceutical companies reduce compliance risks, improve efficiency, and build resilience against regulatory changes.
Successful implementation depends on consistent leadership commitment and a culture of quality where compliance is seen not as a burden but as an enabler of trust and innovation.
- Develop and enforce clear compliance policies: organizations must create written policies that define how electronic records and signatures are handled. These policies provide the framework for daily operations and ensure consistency across departments.
- Train staff regularly: employees must understand both the regulatory requirements and their role in compliance. Regular training reduces human error and reinforces a culture of accountability.
- Conduct periodic system validation and internal audits: compliance is not a one-time effort. Regular validation ensures that systems remain compliant as technology and processes evolve, while audits identify gaps before they escalate into regulatory issues.
- Maintain updated SOPs: Standard Operating Procedures document workflows and responsibilities. Keeping SOPs current ensures that teams always operate within the latest compliance framework and are prepared for inspections.
Ensuring 21 CFR Part 11 compliance
In conclusion, these are the key factors to consider when promoting 21 CFR Part 11 compliance and digital signatures within your organization. In this regulation, the FDA describes specific criteria for electronic records and electronic signatures—specifically for life science companies that export their pharmaceuticals or medical devices to the United States.
The regulation is designed to promote the undisputed approval of signatures, the traceability of changes, and the prevention of falsified records via robust data security measures.
To this end, organizations should always choose tools that come with built in 21 CFR Part 11 compliance such as an eQMS.





